腓骨肌萎縮癥(CMT)1型的臨床、神經電生理及CMT1A型基因診斷方法的研究
2013-09-20 08:03:56曹秉振
中風與神經疾病雜志 2013年3期
關鍵詞:檢測
史 磊, 曹秉振
腓骨肌萎縮癥(charcot-marie-tooth disease,CMT)又稱遺傳性運動感覺神經病(hereditary motor and sensory neuropathy,HMSN),是最常見的遺傳性周圍神經疾病,發病率為1/2500[1]。其主要特征為慢性進行性加重的肢體遠端無力、肌萎縮和感覺損害。CMT的遺傳方式主要為常染色體顯性遺傳(AD),也可見常染色體隱性遺傳(AR)及X連鎖顯性或隱性遺傳(XD或 XR)。根據電生理和病理特征,CMT可分為CMT1(脫髓鞘型)、CMT2(軸索型)和其他較罕見的類型[2]。目前,分子生物學研究已經發現了近40個不同的CMT致病基因位點,20多個致病基因已被克隆。臨床上最常見的類型為CMT1型,其中50% ~70%的CMT 1患者及90%的散發病例為 CMT1A型[3],多由17號染色體短臂11.2區(17p11.2)包含周圍髓鞘蛋白 PMP 22基因在內的1.5MB的正向串聯重復突變所致。
我們收集了5例臨床基本確診為CMT1型的患者,分析比較他們的臨床和電生理特點,并分別應用等位基因特異性PCR和多重連接探針擴增技術(MLPA)兩種方法對5例患者進行PMP22基因重復突變檢測,初步驗證兩種方法所得結果的一致性,指導臨床開展相關基因篩查和診斷的基本程序以進一步確定CMT1A型的診斷。
1 對象與方法
1.1 研究對象 5例CMT1型患者均來源于濟南軍區總醫院神經內科門診及住院的病例。所有患者均由神經內科專科醫生進行詳細的病史詢問和體格檢查,符合Harding于1980年制定的診斷標準[4]。所有受試者均征得本人知情同意。
1.2 神經電生理檢查 主要為運動和感覺神經傳導速度的檢查,上肢包括正中神經和尺神經,下肢運動神經主要檢查腓總神經和脛神經,感覺神經為腓淺神經和腓腸神經。
1.3 基因突變檢測
1.3.1 等位基因特異性PCR檢測 抽取患者外周靜脈血3~5ml,用EDTA抗凝,Promega試劑盒提取全血標本基因組DNA。在PMP22基因重復片段的遠端和近端的區域內,設計出等位基因特異性引物,使其可擴增出患者特異性重復連接片段[5],PCR反應體系為:總體積50μl,其中 dNTP Mixture(2mmol/L)5μl,10 × PCR buffer 5μl,TaqDNA 聚合酶0.25U,基因組 DNA2μl,上下游引物(5μmol/L)各5μl,三蒸水 27.75μl。反應條件為在 GeneAmp PCR system 2400熱循環儀上94℃預變性5min,然后 94℃變性 30s,56℃退火 1min,72℃延伸 3min,25次循環,最后72℃延伸5min,4℃保存。反應完成后取5μl PCR產物與1μl溴乙錠混合,0.8%瓊脂糖凝膠電泳(加核酸染色液)檢測產物。
1.3.2 MLPA檢測 MLPA檢測試劑盒 SALSA P033 購自 MRC-Holland,取基因組 DNA 1μl,加TE稀釋至5μl,在熱循環儀上98℃變性5min后冷卻至 25℃,分別加入 1.5μl P033探針混合物及1.5μl MLPA buffer,小心混勻后 95℃ 變性 1min,60℃下雜交16h~20h。雜交后在54℃下加入連接混合物(25μl三蒸水,3μl連接緩沖液 A,3μl連接緩沖液 B,1μl連接酶)孵育15min,然后98℃加熱5min以滅活連接酶,待溫度降至20℃后,加入PCR反應混合物(7.5μl三蒸水,2μlSALSA PCR 引物混合物,0.5μl SALSA 聚合酶),PCR 反應條件為:95℃變性30s,60℃ 退火 30s,72℃ 延伸 60s,循環 35 次,72℃延伸 20min,取0.7μl PCR 反應產物加入 0.2μl LIZ-500標記物及9μl甲酰胺(LIZ-500及甲酰胺均為美國ABI公司生產),80℃變性2min后立即置冰架上冷卻,最后用毛細管凝膠電泳檢測(在ABI3130基因測序儀上進行),程序結束后導出數據,結果用Coffalyser軟件分析。
2 結果
2.1 臨床特征 4例患者20歲前發病,其中2例有家族史,1例患者30歲左右發病,無家族史。所有患者均表現進行性四肢遠端無力伴肌萎縮,下肢重于上肢,4例患者四肢遠端感覺減退,1例患者感覺減退不明顯。所有患者均出現腱反射減弱或消失,2例患者出現足弓變形。
2.2 神經電生理檢查 2例患者上肢正中神經傳導速度嚴重降低(<25m/s),3例患者上肢正中神經傳導速度明顯降低(<38m/s),4例患者下肢腓總神經傳導速度嚴重降低(<20m/s),1例患者明顯降低(<38m/s)。4例患者所檢上下肢感覺神經未引出動作電位,1例患者所檢感覺神經傳導速度明顯降低。患者所檢神經波幅均有不同程度降低。
2.3 基因突變分析
2.3.1 等位基因特異性PCR產物檢測結果1例患者出現了經特異引物擴增的特異性短片段重復的異常條帶,其余4例患者未出現異常條帶。
2.3.2 MLPA檢測分析 運行軟件Coffalyser分析得出:4例患者的PMP22基因全部在正常范圍內,只有1例患者PMP22基因的幾乎所有外顯子均在正常范圍上限以上,表明這例患者存在PMP22基因的重復突變(圖1為PMP22基因重復突變患者的分析結果,正常范圍結果圖略)。
3 討論
CMT1型的患者發病年齡較早,常兒童期起病,患者表現比同齡兒童跑跳困難,而后出現進行性四肢無力和肌萎縮,嚴重時可見爪型手和弓形足。此次收集的病例臨床癥狀相對單一,未見有其他報道中提到的神經功能重度受損、,如伴錐體束征、伴神經性耳聾等相關癥狀出現[6],說明CMT1臨床表型的變異較大。
臨床上常根據電生理和病理特點大致分為CMT1型(脫髓鞘型)和CMT2型(軸索型),電生理主要依據神經傳導速度(NCV)減慢的程度和波幅是否降低未區分,脫髓鞘型一般表現為正中神經NCV<38m/s且波幅無明顯降低,而軸索型出現波幅明顯降低但正中神經NCV >38m/s。本研究中患者均出現運動神經傳導速度減慢,其中2例較嚴重病例正中神經NCV<25m/s,下肢腓總神經NCV<20m/s,且出現繼發性波幅降低。患者雖表現四肢末端感覺障礙較輕但是感覺神經的動作電位較難引出。可見神經傳導速度降低往往會繼發波幅降低,同樣波幅降低也會繼發神經傳導速度減慢,所以很多情況根據電生理不好明確區分。神經病理活檢可以明確患者為何種類型,但損傷較大,患者接受程度較低。相比之下基因檢測的準確性高,基本無損傷,在病程早期即可做出明確診斷,有較高的臨床應用價值。
本研究中采用了兩種方法檢測患者的PMP22基因是否存在短片段重復突變,等位基因特異性PCR是直接對PMP22基因交換熱點區設計引物擴增出特異性連接片段,Kon-Ping Lin等根據已被共識的近端和遠端CMT1A-REP序列設計出了PMP22等位基因特異性引物[7],其優點是簡便易行,對操作條件及成本要求較低,但其靈敏度和準確性仍需進一步驗證。而新技術MLPA-CMT1A試劑盒的探針基本覆蓋了PMP22基因外顯子的所有熱點區域,且能同時檢測基因的重復與缺失,敏感度及準確度較等位基因特異性PCR更高,且MLPA與熒光定量PCR、STR等方法相比也具有一定的優勢:(1)可同時檢測多基因多外顯子的重復或缺失。(2)對DNA模板定量及純度要求較低。當然,MLPA也有自身的缺點和限制,比如價格稍昂貴、對實驗室的配置條件要求較高等。因此,兩種方法對特異性短片段重復突變的檢測都具有不可替代的作用和意義。建議在臨床患者初篩或基礎設施比較簡單的實驗室條件下,可以先采用等位基因特異性PCR進行檢測,對可疑陽性患者再進一步選擇靈敏度和準確度更高的MLPA確定診斷。
CMT涉及的致病基因較多,僅CMT1型相關的基因就有6個,基因檢測雖然準確性高但要確定具體的致病基因可能需要多次實驗,未檢測出PMP22基因重復突變的患者很有可能存在其他基因的突變,需要進一步實驗驗證。本實驗綜合研究了CMT1型患者的臨床和電生理特征,同時研究了CMT1A型基因突變的檢測方法,旨在指導臨床根據電生理特征及PMP22基因重復突變的檢測更好地確定CMT1A型的診斷。
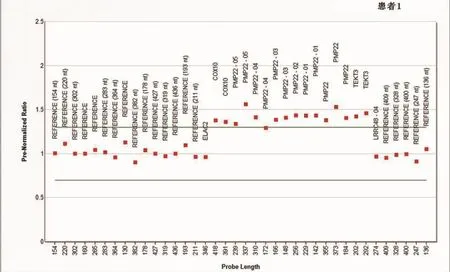
圖1 PMP22基因重復突變患者的分析結果
[1]Berger P,Young P,Suter U,etal.Molecular cell biology of Charcot-Marie-Tooth disease[J].Neurogenetics,2002,4(1):1-15.
[2]Street VA,Bennett CL,Goldy JD.Mutation of a putative protein degradation gene LITAG/SIMPLE in Charcot-Marie-Tooth disease LC[J].Neurology,2003,60:22.
[3]蕭劍鋒,唐北沙,夏家輝,等.聚合酶鏈反應技術在腓骨肌萎縮癥基因診斷中的應用[J].中華醫學雜志,2001,81:138.
[4]Harding AE,Thomas PK.The clinical features of hereditary motor and sensory neutopathy types1 and 2[J].Brain,1980,103(2):258-280.
[5]Lin KP,Chou CH,Lee HY,etal.Allele-specific.All-or-None PCR product diagnostic strategy for Charcot-Marie-Tooth 1A and hereditary neuropathy with liability to pressure palsies[J].JChin Med Assoc,2006,69(2):68-73.
[6]郭 鵬,宋福聰,王相斌,等.常染色體顯性遺傳腓骨肌萎縮癥的臨床與基因突變特點[J].中風與神經疾病雜志,2011,28(8):705-707.
[7]Zmurovic N,Millc V,Dackovic J,etal.Analysisi ofmutation in the chromosome 17p11.2 region in patients with Charcot-Marie-Tooth type 1 disease and in patients with tomaculous neuropathy[J].Srp Arh Celok Lek,2002,130(3-4):59-63
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
