Aβ25-35寡聚體對大鼠海馬神經細胞的活性影響及形態學觀察
2013-09-20 08:03:56黃昕艷趙錦程尉榮翠朱曉峰
中風與神經疾病雜志 2013年3期
黃昕艷, 趙錦程, 尉榮翠, 楊 智, 劉 爽, 朱曉峰
阿爾茨海默病(Alzheimer’s disease,AD)是一種慢性進行性神經退變性病變,其典型的臨床病理學改變為β淀粉樣蛋白(β-amyloid protein,Aβ)沉積、神經元纖維纏結和神經元缺失[1]。Aβ是老年斑的核心成分,在AD的發生、發展過程中起關鍵作用,本實驗旨在通過Aβ25-35寡聚體誘導海馬神經元細胞損傷來探討其活性及形態學指標的變化,篩選Aβ25-35寡聚體致傷條件下適宜的AD細胞模型。
1 材料與方法
1.1 材料 (1)動物:新生 Wistar大鼠(1~3d),由佳木斯大學實驗動物中心提供。(2)主要試劑:DMEM/F12干粉培養基、B27、Neurobasal-A培養基、胎牛血清、胰蛋白酶(購于美國Gibco公司);Aβ25-35、二甲基亞砜(DMSO)、四甲基偶氮唑鹽(MTT)、HFIP(六氟丙醇)購于武漢博士德公司;兔抗大鼠神經元特異性烯醇化酶(neuron specific enolase,NSE)單克隆抗體、SABC即用型試劑盒、DAB顯色試劑盒(購于Boster公司);青霉素、鏈霉素(華北制藥股份有限公司)。
1.2 方法
1.2.1 原代海馬神經元培養 出生1~3d內的Wistar大鼠,無菌條件下斷頭,在冰浴的D-hank s液中取腦并分離出雙側海馬組織,剪切成約1mm×1mm×1mm的組織塊,0.125%胰酶37℃消化約20min。用含體積分數為10%胎牛血清的DMEM/F12培養基中終止消化,用口徑光滑的小吸管反復吹打分散,200目細胞篩過濾后,1000r/min,離心5min,棄掉上清液,加入培養基重懸,制成(2~3)×105個/ml密度的細胞懸液,接種于預先用0.1mg/ml多聚賴氨酸包被的96、24、6孔板中。置于37℃、5%CO2培養箱內培養,24h后換成Neurobasl+B27(2%)培養基,以后每3d換液1次。
1.2.2 神經元鑒定 培養至7d的神經細胞,以PBS輕洗3遍,4%多聚甲醛固定30min后,PBS洗2次。0.4%TritonX-100破膜 20min,PBS洗 3遍,山羊血清封閉30min,滴加NSE(1∶200稀釋)一抗,4℃濕盒內孵育過夜。次日PBS洗3遍,加二抗室溫避光孵育50min,PBS洗3遍;加新鮮配制的DAB溶液顯色,待顯色充分后,加入三蒸水終止反應,梯度乙醇脫水,二甲苯透明,中性樹膠封片,光鏡觀察,(PBS代替一抗做陰性對照)。
1.2.3 實驗分組 將原代培養8d后的神經元隨機分為5組:正常對照組、損傷組,加入終濃度分別為 1.0、5.0、10.0、20.0μmol/L 的 Aβ25-35寡聚體,Aβ25-35寡聚體的制備參考王建秀等文獻[2],繼續培養24h。
1.2.4 神經細胞形態學觀察和定量分析Aβ25-35寡聚體孵育24h后,于倒置顯微鏡下觀察其存活情況和形態,隨機選取30個視野,記錄每個視野內有突起的細胞數和突起總數,同時目鏡微尺測量突起的長度,重復3次。
1.2.5 MTT法檢測細胞活性 采用MTT比色法檢測培養細胞的存活率。按上述分組處理24h后,向96孔板中各組細胞分別加入 5mg/ml MTT20μl,37℃繼續培養4h后棄上清液,每孔加入150μl DMSO,振蕩10min,待紫色結晶完全溶解后,用酶標儀檢測各組細胞570nm吸光度值(opti-cal density,OD)。每個濃度設5個復孔,計算其平均值,同時設不加細胞的空白孔。根據公式計算:細胞存活率(%)=處理組細胞OD值/對照組細胞OD值×100%。
2 結果
2.1 神經元形態學觀察及鑒定 接種后的海馬神經細胞呈圓形,單個均勻分布,2h后開始貼壁,培養24h后細胞大部貼壁且部分已長出突起。隨培養時間延長,突起逐漸增多、延長并交織成網。胞體呈多角形或橢圓形,周圍有光暈,胞體體積較大,透光性強,核大;培養至7d的細胞胞體飽滿、有立體感;突起明顯增多、增長、交織成網、細胞生長成熟。與陰性對照、陽性對照比較,NSE免疫染色顯示95%以上的培養細胞為陽性染色,符合細胞原代培養純度要求(見圖1)。

圖1 神經細胞原代培養(×100)
Aβ致傷模型組可見到部分神經細胞出現胞體縮小,細胞周圍暈光不強;細胞突起消失或明顯減少、縮短,細胞碎片增多,甚至細胞崩解,神經細胞數目下降等變化。Aβ1.0μmol/L組神經細胞形態基本正常,貼壁良好、突起較長,可相互連接成網絡。Aβ5.0μmol/L組神經細胞生長狀態不如正常對照組旺盛,神經元突起數目、長度與正常對照組相比有明顯的差異(P <0.05,P <0.01)。Aβ10.0μmol/L組神經細胞存活狀態欠佳,突起變短,有較多的圓形及浮細胞,其突起的細胞數、神經元突起數目、長度與正常對照組相比有顯著的差異(P<0.05,P<0.01,P < 0.01)。Aβ20.0μmol/L 組神經細胞生長較差,多數細胞突起變短、消失,細胞碎片較多。其突起的細胞數、神經元突起數目、長度與正常對照組相比有顯著的差異(P <0.01,P <0.01,P <0.01),見圖2、表1。
2.2 Aβ25-35寡聚體誘導損傷后神經細胞存活的影響 MTT測定結果顯示,Aβ25-35寡聚體可導致神經細胞抑制,且神經細胞抑制表現為與Aβ寡聚體濃度依賴性關系,即隨著Aβ寡聚體濃度增加神經細胞,存活率逐漸下降,其中20μmol/L組最明顯,與空白對照組相比,差異有統計學意義(P<0.05);Aβ25-35寡聚體毒性會導致細胞損傷(見圖2)。
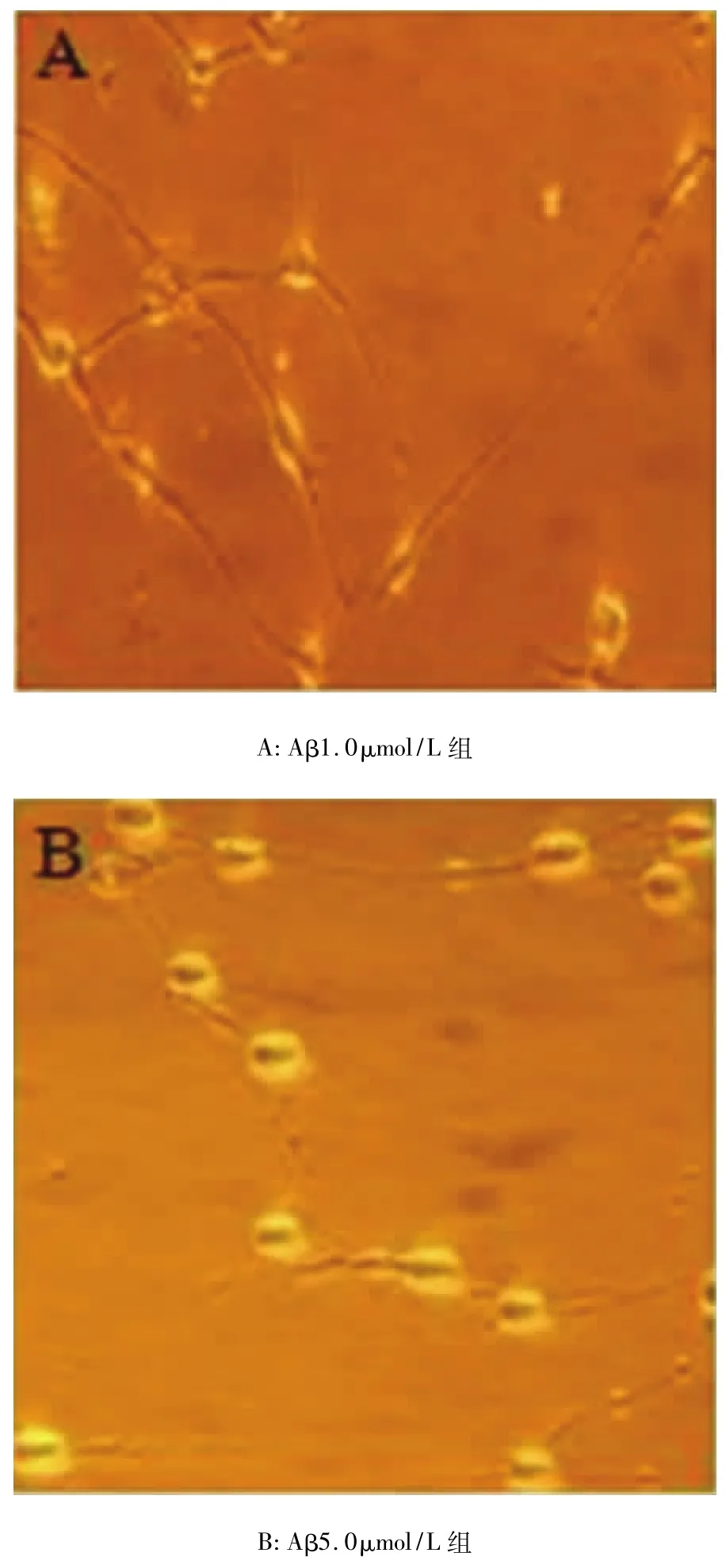
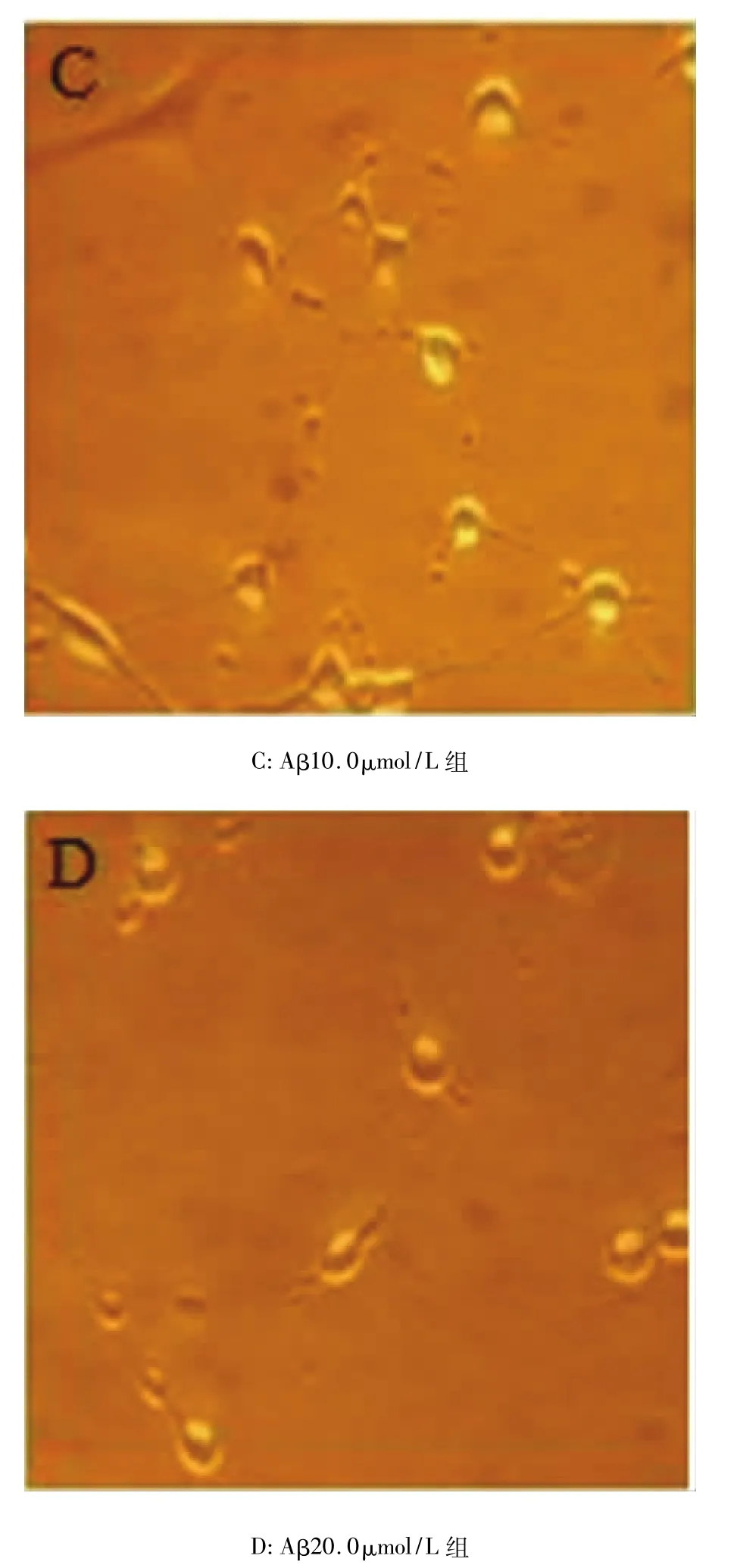
圖2 Aβ不同致傷濃度對原代培養神經細胞的影響(倒置顯微鏡,×100)
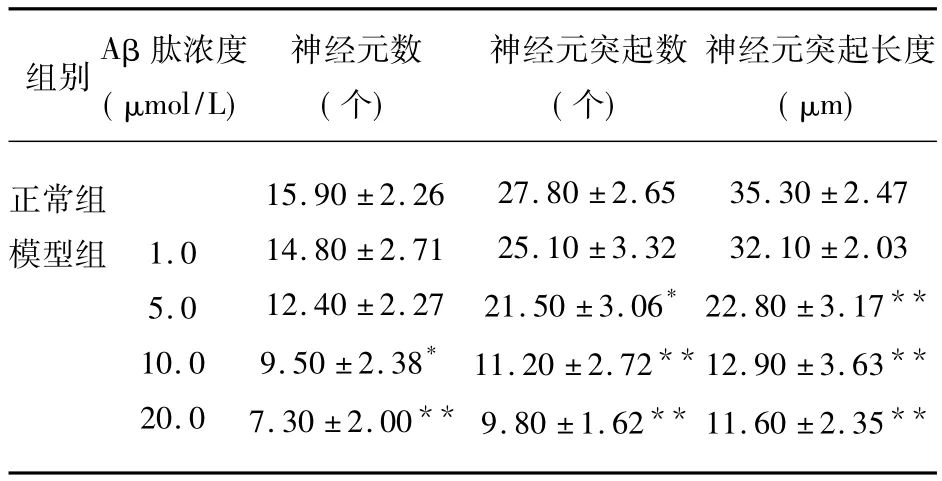
表1 Aβ25-35對神經細胞形態學改變的影響(χ ± s,n=30)
表2 不同濃度Aβ肽對海馬神經元的影響(±s)
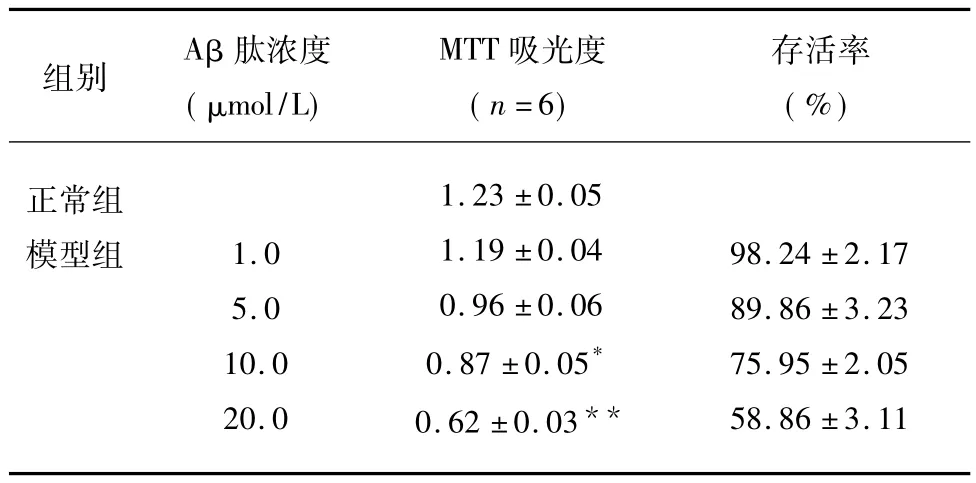
表2 不同濃度Aβ肽對海馬神經元的影響(±s)
與空白對照組比較*P <0.05,**P <0.01
組別Aβ肽濃度(μmol/L)MTT吸光度(n=6)存活率(%)正常組模型組 1.0 5.0 10.0 20.0 1.23 ±0.05 1.19 ±0.04 0.96 ±0.06 0.87 ±0.05*0.62 ±0.03**98.24 ±2.17 89.86 ±3.23 75.95 ±2.05 58.86 ±3.11
3 討論
Aβ級聯學說的提出源自對AD的神經病理特征的發現,已經確認老年斑的主要構成物質是β淀粉樣物質,Aβ在腦內異常聚集引起神經毒性,包括直接毒性和增強、放大各種傷害性毒性,導致神經細胞死亡[3]。
目前認為,Aβ在大腦皮質內的蓄積是AD病理發生過程中的一個早期必然事件[4],研究表明,Aβ在形成纖維性沉積前的中間體狀態,即Aβ寡聚體亦可產生神經毒性作用。動物實驗研究表明,Aβ寡聚體可以在生理水平明顯抑制大鼠海馬的突觸傳遞[5]。且發現在AD發病早期還未出現明顯的老年斑之前,其認知功能的減退與可溶性Aβ水平及突觸功能障礙密切相關[6,7]。因此,建立良好而穩定的AD突觸損傷模型對探索AD早期記憶損害事件的成因及AD發病機制的研究非常重要。
Aβ肽全長的第25~35個氨基酸殘基是Aβ的非生理性最小毒性片段,故本實驗以Aβ25-35寡聚體致傷與記憶密切相關的海馬神經元為研究對象,來篩選適宜的AD突觸損傷模型,為AD早期的防治探求的有效的途徑。
Aβ25-35寡聚體致傷 24h后從形態學觀察,Aβ25-35寡聚體可致神經細胞損傷,且神經細胞毒性表現與 Aβ25-35寡聚體呈劑量依賴性關系,其中Aβ25-3520μmol/L組致傷顯著,細胞存活狀態差,有部分細胞脫壁,細胞間網絡消失,細胞外基質雜亂,其突起的細胞數、神經元突起數目、長度與正常對照組相比有顯著的差異(P<0.01);不適宜作為AD突觸損傷模型進行下一步研究。
Aβ25-3510μmol/L組突起的細胞數、神經元突起數目與Aβ25-3520μmol/L組相比有所改善,突起的細胞數與正常對照組相比有一定差異(P<0.05),但神經元突起數及突起的長度與正常對照組相比仍有顯著的差異(P<0.01),大部分神經細胞突起已喪失,故也不宜作為適宜的突觸損傷模型。
Aβ25-355μmol/L組細胞生存狀態尚可,突起的細胞數與正常對照組比照差異不顯著,神經元突起數目與正常對照組相比(P<0.05),突起的長度與正常對照組相比有顯著的差異(P<0.01),損傷模型中神經細胞數量尚穩定,神經元突起數目有部分損失,但尚可作為Aβ致突觸損傷模型做進一步研究。
對于Aβ25-351μmol/L組細胞損傷效果不明顯,神經元突起數、突起長度與正常對照組比較差異不顯著,不宜作為有效的突觸損傷模型。
在MTT測定結果顯示 Aβ25-3510μmol/L組與Aβ25-3520μmol/L組細胞存活率降低較明顯,不適宜作為AD的突觸損傷模型,同樣支持上述結果。
本實驗以Aβ25-35寡聚體致傷海馬神經細胞,篩選出5μmol/L為適宜的AD突觸損傷模型的致傷濃度,希望為AD早期的防治探索更有效的途徑。
[1]Pereira C,Ferreiro E,Cardoso SM,etal.Cell degeneration induced by amyloid-beta peptides:implications for Alzheimer’s disease[J].Mol Neurosci,2004,23(1):97-104.
[2]王建秀,王德生,段淑榮.Aβ25-35對PC12細胞損傷作用的研究[J].中風與神經疾病雜志,2007,24(2):148.
[3]Lambert JC,Amouyel P.Genetics of Alzheimer’s disease:new evidences for an old hypothesis[J].Curr Opin Genet Dev,2011,21(3):295-301.
[4]Wang HW,Pasternak JF,Kuo H,etal.Soluble oligomers of beta amyloid(1-42)inhibit long-term potentiation but not long-term depression in rat dentate gyrus[J].Brain Res,2002,924:133-140.
[5]Lacor PN,Buniel MC,Chang L,etal.Synaptic targeting by Alzheimer’s-related amyloid beta oligomers[J].J Neurosci,2004,24:10191-10200.
[6]Tannenberg RK,Scott HL,Tannenberg AE,etal.Selective loss of synaptic proteins in Alzheimer’s disease:evidence for an increased severity with APOE varepsilon4[J].Neurochem Int,2006,49(7):631-639.
[7]Spencer B,Rockenstein E,Crews L,etal.Novel strategies for Alzheimer’s disease treatment[J].Expert Opin Biol Ther,2007,7(12):1853-1867.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
音樂探索(2022年2期)2022-05-30 21:01:37
作文周刊·小學二年級版(2022年20期)2022-05-05 01:33:06
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
創新作文(小學版)(2019年10期)2019-09-25 08:12:28
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
小學生學習指導(低年級)(2017年5期)2017-05-04 04:14:38
光學精密工程(2016年6期)2016-11-07 09:07:19
