中國(guó)先天性副肌強(qiáng)直一家系致病基因初步研究
2013-10-20 06:47:37桑道乾談昕煜陳靜時(shí)鵬許文芳陳齊鳴胡松年
中國(guó)實(shí)用醫(yī)藥 2013年20期
桑道乾 談昕煜 陳靜 時(shí)鵬 許文芳 陳齊鳴 胡松年
中國(guó)先天性副肌強(qiáng)直一家系致病基因初步研究
桑道乾 談昕煜 陳靜 時(shí)鵬 許文芳 陳齊鳴 胡松年
目的對(duì)收集的中國(guó)先天性副肌強(qiáng)直(PC)一家系致病基因進(jìn)行初步篩查。方法采用測(cè)序的方法對(duì)中國(guó)PC家系的患者和正常人的SCN4A基因24個(gè)外顯子測(cè)序,分析比較中國(guó)PC家系的患者和正常人與正常人群的差異。結(jié)果未發(fā)現(xiàn)該中國(guó)PC家系在SCN4A基因24個(gè)外顯子存在致病突變。結(jié)論該中國(guó)PC家系的發(fā)病與SCN4A基因無關(guān)。
先天性副肌強(qiáng)直(paramyotonia congenita,PC)又稱為Eulenburg病,是一常染色體顯性遺傳病,以冷刺激或運(yùn)動(dòng)誘發(fā)的發(fā)作性面、舌、頸和手部肌強(qiáng)直,持續(xù)數(shù)分至數(shù)小時(shí)為顯著特征,臨床罕見。目前多數(shù)學(xué)者認(rèn)為是骨骼肌細(xì)胞膜上鈉通道Ⅳ型α亞單位(alpha subunit type Ⅳ of voltage-gated sodium channel,SCN4A)基因突變所致。本研究收集了中國(guó)一家族性先天性副肌強(qiáng)直,調(diào)查和分析患者的臨床特征,并對(duì)其致病基因進(jìn)行了初步篩查。
1 家系資料
調(diào)查該家系3代,有血緣關(guān)系者15人,如下圖。明確發(fā)病者2代4人即:
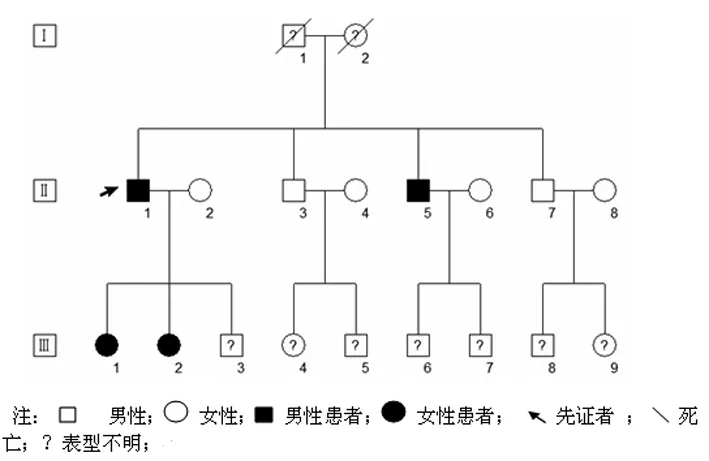
Ⅱ1、Ⅱ5、Ⅲ1、Ⅲ2、Ⅲ3、Ⅲ4、Ⅲ5、Ⅲ6、Ⅲ7、Ⅲ8和Ⅲ9因年齡小尚不明確是否為患者。Ⅰ1和Ⅰ2因死亡年代久遠(yuǎn),無從知曉是否為患者。先證者Ⅱ1,男,60歲,電工。發(fā)作性肌肉僵硬三十年入院。三十年前出現(xiàn)雙下肢小腿和大腿肌肉僵硬,常在運(yùn)動(dòng)后或勞累后發(fā)作,發(fā)作時(shí)持續(xù)一到數(shù)分鐘,數(shù)天發(fā)作一次。肌肉僵硬逐漸累及腹肌、胸背肌肉、雙上肢肌肉、頸部肌肉和咀嚼肌肉。常有張口后不能立刻閉觜,雙手長(zhǎng)時(shí)用力持握老虎鉗或冬季騎摩托車不能放松,癥狀持續(xù)約5 min后緩解。同時(shí)有夜間起床后不能立即行走,需全身?yè)u晃5~10 min后才能行走。肌肉僵硬冬天較夏天發(fā)作頻繁。近兩月發(fā)作加重,一天數(shù)次,累及腹肌、胸背肌肉、四肢肌肉、頸部肌肉和咀嚼肌肉。患者2女兒和一弟有類似癥狀。體檢:寬矮體型,四肢肌力和肌張力正常,深反射正常,病理反射未引出,腓腸肌、肱二頭肌叩診未見肌球;右手冰水侵浴10min后見肌肉僵硬發(fā)作。輔助檢查:電解質(zhì)結(jié)果正常,肌酶譜結(jié)果正常;肌電圖檢查未見強(qiáng)直放電;重復(fù)刺激實(shí)驗(yàn)未見運(yùn)動(dòng)復(fù)合電位(compound motor action potential, CMAP)波幅明顯衰減;短時(shí)運(yùn)動(dòng)誘發(fā)電位實(shí)驗(yàn)可見CMAP波幅即刻輕微衰減,長(zhǎng)時(shí)運(yùn)動(dòng)誘發(fā)電位實(shí)驗(yàn)可見CMAP波幅延遲輕微衰減,運(yùn)動(dòng)后半小時(shí)CMAP波幅達(dá)最低點(diǎn),然后緩慢恢復(fù),90分鐘后恢復(fù)初始波幅。苯妥英鈉,0.1g/次,3次/d無效,0.2g/次,3次/d,每天三次有效。
該家系患者共同的臨床特征是25歲后發(fā)病,寒冷和運(yùn)動(dòng)誘發(fā)肌肉僵硬,苯妥英鈉治療有效。先證者Ⅱ1特征性的癥狀是雙手長(zhǎng)時(shí)用力持握老虎鉗或冬季騎摩托車不能放松。先證者Ⅱ1的女兒Ⅲ1和Ⅲ2特征性的癥狀是雙手常在冬季洗衣時(shí)出現(xiàn)僵硬。先證者Ⅱ1的弟弟Ⅱ5是搬運(yùn)工,特征性的癥狀是搬運(yùn)貨物時(shí)常出現(xiàn)四肢僵硬。
2 分子遺傳學(xué)研究
對(duì)家系中表型明確的患者和正常人進(jìn)行SCN4A基因24個(gè)外顯子測(cè)序,分析比較家系中患者和正常人以及正常人群的差異。
2.1DNA提取 采集患者Ⅱ1、Ⅱ5、Ⅲ1、Ⅲ2和家系中正常人Ⅱ3、Ⅱ7靜脈血2毫升,使用Axygen血基因組DNA小量試劑盒提取DNA。
2.2SCN4A基因24個(gè)外顯子PCR擴(kuò)增及測(cè)序引物序列如下表。

表1
2.3PCR擴(kuò)增體系及條件 PCR擴(kuò)增體系(15μl): DNA模板1.0μl,10xBuffer(Mg2+) 1.5μl,正向引物(10 mM)0.3μl,負(fù)向引物(10 mM)0.3μl, dNTP (2.5 mM)0.4μl,Taq酶 (5U/μl)0.3μl,ddH2O 11.1μl。PCR擴(kuò)增條件:94°C 預(yù)變性3 min,94°C變性 30s,56°C復(fù)性30s,72°C 延伸1 min,共32個(gè)循環(huán),最后72°C延伸10 min。
2.4AxyPrep DNA 凝膠回收試劑盒純化PCR產(chǎn)物。
2.5測(cè)序反應(yīng) (1)測(cè)序PCR反應(yīng)體系(10μl): 純化的PCR產(chǎn)物2μl,測(cè)序引物NF 1μl,BigDye 2μl,ddH2O 5μl。(2)測(cè)序PCR擴(kuò)增條件:96℃變性2 min后進(jìn)行PCR循環(huán),PCR循環(huán)參數(shù)為96℃ 10s,50℃ 5s,60℃3Min10s,30個(gè)循環(huán)。(3)測(cè)序反應(yīng)的純化后上測(cè)序儀ABI 3730XL測(cè)序儀測(cè)序。
2.6序列變異分析結(jié)果 家系中正常人和患者SCN4A基因的第10外顯子的變異位點(diǎn)為第118位核苷酸由腺嘌呤核苷酸變異為鳥嘌呤核苷酸,導(dǎo)致其所編碼的氨基酸由Ser(絲氨酸)變?yōu)镚ly(甘氨酸),單核苷酸多態(tài)(SNP)編號(hào)為rs6504191,該核苷酸位于轉(zhuǎn)錄本NM000334的第1647位。家系中正常人和患者SCN4A基因的其他外顯子未發(fā)現(xiàn)變異。
3 討論
PC的顯著癥狀是寒冷或運(yùn)動(dòng)誘發(fā)的骨骼肌強(qiáng)直,與先天性肌強(qiáng)直不同的是PC無運(yùn)動(dòng)后強(qiáng)直緩解所謂溫?zé)岈F(xiàn)象(warm-up phenomenon)。診斷PC主要依靠臨床特征[1]。肌強(qiáng)直放電有助于診斷,但肌強(qiáng)直放電受溫度影響。當(dāng)溫度低于28°C時(shí),肌電圖檢查會(huì)出現(xiàn)纖顫電位;當(dāng)溫度低于20°C時(shí),肌強(qiáng)直性放電消失[2]。有些患者,室溫下肌電圖檢查可完全正常[3]。因此,依靠肌電圖所見肌強(qiáng)直放電診斷PC不可靠。本家系肌電圖檢查未出現(xiàn)肌強(qiáng)直性放電可能是檢查條件限制所致。本家系臨床特征符合PC診斷條件,短時(shí)和長(zhǎng)時(shí)運(yùn)動(dòng)誘發(fā)肌電圖也支持診斷。
目前對(duì)于PC的分子遺傳學(xué)研究文獻(xiàn)報(bào)道集中于SCN4A基因,該基因位于17q23-q25.3。Park JH等報(bào)道一韓國(guó)PC家系患者的致病突變位點(diǎn)為SCN4A G1306V[4]。Koch MC等[5]報(bào)道一德國(guó)PC家系患者的致病突變位點(diǎn)為SCN4A Val1293Ile。然而Schose BGH等[6]對(duì)另一德國(guó)4代17例患者的PC大家系研究發(fā)現(xiàn)致病位點(diǎn)是SCN4AA1481D。Davies NP等[7]研究發(fā)現(xiàn)一愛爾蘭PC家系的致病突變是SCN4AG4367A。Sallansonnet-Froment M等[8]報(bào)道法國(guó)一PC家系的致病位點(diǎn)在SCN4AArg1448Cys。Kinoshita M等[9]報(bào)道日本一PC家系的致病位點(diǎn)在SCN4A Thrl313Met。Parasivam S等[10]發(fā)現(xiàn)澳大利亞PC家系的致病位點(diǎn)在SCN4AT1313。Feng Y等[11]研究發(fā)現(xiàn)中國(guó)伴有周期性麻痹的PC家系的突變位點(diǎn)在SCN4AMet1592Val。Matthew E等[12]對(duì)來自英國(guó)的42例PC患者,其中包括28例有血緣關(guān)系的PC患者進(jìn)行研究,結(jié)果發(fā)現(xiàn)R1448L 和L1436P兩個(gè)新的致病位點(diǎn),同時(shí)也發(fā)現(xiàn)10例(包括8例有血緣關(guān)系)PC無SCN4A無突變。Sampaolo S等[13]報(bào)道一意大利PC家系的SCN4A基因既無突變也無異常剪切。由此可見,PC存在遺傳異質(zhì)性。本研究對(duì)SCN4A基因的24個(gè)外顯子測(cè)序未發(fā)現(xiàn)致病突變,初步排除了SCN4A基因?yàn)樵撝袊?guó)PC家系的致病基因。該中國(guó)PC家系的致病基因有待進(jìn)一步研究。
[1] Heatwole CR, Moxley III RT. Journal of the American Society for Experimental NeuroTherapeutics (Neurotherapeutics), 2007,4: 238-251.
[2] Shapiro B, Ruff R. Disorders of skeletal muscle membrane excitability:myotonia congenita, paramyotonia congenita, periodic paralysis,and related disorders. In: Katirji B, Kaminski HJ, Preston DC,Ruff RL, Shapiro BE, editors. Neuromuscular disorders in clinical practice. Boston: Butterworth-Heinemann; 2002:987-1020.
[3] Russell SH, Hirsch NP. Anaesthesia and myotonia. Br J Anaesth 1994;72:210 -216.
[4] Park JH, Lee YW, Park SA, et al. A Case of Paramyotonia Congenita Without Periodic Paralysis Electrophysiological and Molecular Genetic Studies. The Neurologist, 2010, 16: 203-205.
[5] Koch MC, Baumbach K, George AL, Ricker K. Paramyotonia congenita without paralysis on exposure to cold: a novel mutation in the SCN4A gene (Val1293Ile). Neuroreport, 1995, 6(15): 2001-2004.
[6] Schose BGH, Schrd O¨er JM, Grimm T,et al. ALARGE GERMAN KINDRED WITH COLD-AGGRAVATED MYOTONIA AND A HETEROZYGOUS A1481D MUTATION IN THE SCN4A GENE. Muscle Nerve, 2007, 35: 599-606.
[7] Davies NP, Eunson LH, Gregory RP, et al.Clinical, electrophysiological, and molecular genetic studies in a new family with paramyotonia congenital. J Neurol Neurosurg Psychiatry, 2000, 68:504-507.
[8] Sallansonnet-Froment M, Bounolleau P, Greslan TD,et al. Paramyotonie congénitale d’Eulenburg. Rev Neurol (Paris), 2007,163 (11) :1083-1090.
[9] Kinoshita M, Sasaki R, Nagano T, et al.Thrl313Met Mutation in Skeletal Muscle Sodium Channels in a Japanese Family with Paramyotonia Congenita. Internal Medicine, 2003, 42,856-861.
[10] Parasivam S, Krupa M, Slee M, et al.Clinical, electrophysiological and genetic features of a large Australian family with paramyotonia congenital. MJA, 2009,190: 334-336.
[11] Feng Y, Ji XP, Sun XH,et al.Severe phenotypes of paralysis periodica paramyotonia are associated with the Met1592Val mutation in the voltage-gated sodium channel gene (SCN4A) in a Chinese family. Journal of Clinical Neuroscience, 2011,18: 1138-1140.
[12] Matthews E, Tan SV, Fialho D,et al.What causes paramyotonia in the United Kingdom? Neurology,2008,70:50-53.
[13] Sampaolo S, Puca AA, Nigro V, et al. Lack of sodium channel mutation in an Italian family with paramyotonia congenita. Neurology, 1999, 53:1549-1555.
PreliminarystudyofcausativegeneforaChinesefamilyparamyotoniacongenital
SANGDao-qian,TANXin-yu,CHENJing,etal.
DepartmentofNeurology,TthefirstAffiliatedHospitalofBengbuMedicalCollege,AnhuiBengbu233004,China
ObjectiveTo study the causative gene in a Chinese family paramyotonia congenital(FPC).MethodsDNA sequencing analysis for exons of alpha subunit type Ⅳ of voltage-gated sodium channel (SCN4A) was conducted between affected and normal individuals of the Chinese FPC and other normal people, each other.ResultsUnexpectly, no causative mutation for the Chinese FPC was found in SCN4A.Conclusionthe Chinese FPC has no relationship with SCN4A.
233004 蚌埠醫(yī)學(xué)院第一附屬醫(yī)院神經(jīng)內(nèi)科(桑道乾 陳靜 時(shí)鵬 許文芳 陳齊鳴);中國(guó)科學(xué)院北京基因組研究所(談昕煜 胡松年)
