沉默Notch1基因促進人乳腺癌MCF-7細胞JNK1和p53磷酸化*
2013-10-24 08:22:45陳旭東范文娟楊旭光王建國
中國病理生理雜志 2013年6期
關鍵詞:信號
袁 磊, 陳旭東, 范文娟, 楊旭光, 王建國
(漯河醫學高等專科學校,河南 漯河 462002)
沉默Notch1基因促進人乳腺癌MCF-7細胞JNK1和p53磷酸化*
袁 磊, 陳旭東, 范文娟, 楊旭光, 王建國△
(漯河醫學高等專科學校,河南 漯河 462002)
目的探究沉默Notch1基因對人乳腺癌MCF-7細胞JNK1和p53磷酸化的影響。方法選取人乳腺癌MCF-7細胞作為研究對象,構建shRNA-Notch1真核表達質粒用于轉染MCF-7細胞使Notch1基因沉默。采用Western blotting方法檢測MCF-7細胞Notch1、Hes-1、PUMA和NOXA蛋白的表達,JNK1和p53蛋白磷酸化水平以及caspase-3活化水平的改變。應用流式細胞術檢測細胞凋亡和線粒體膜電位的變化。結果人乳腺癌MCF-7細胞Notch1基因被沉默后,Notch1和Hes-1蛋白表達量明顯減少(P<0.01),細胞凋亡率顯著升高(P<0.01),JNK1和p53的磷酸化水平明顯高于對照組(P<0.01),PUMA和NOXA表達量顯著升高(P<0.05),cleaved caspase-3蛋白明顯多于對照組(P<0.01),線粒體膜電位明顯下降(P<0.05)。結論沉默Notch1基因可能通過激活JNK1信號通路活化p53,促進PUMA和NOXA蛋白表達,進而通過線粒體途徑導致人乳腺癌MCF-7細胞凋亡。
Notch1蛋白; 短發夾RNA; JNK1蛋白; p53蛋白; MCF-7細胞
近年來,在果蠅發育的研究中,發現了一條在細胞間傳導相互作用的信號途徑,稱為Notch信號途徑。隨后證實該信號途徑廣泛存在于無脊椎動物和脊椎動物體內,并且在進化過程中高度保守。Notch信號途徑的激活始于Notch受體胞外區與相鄰細胞表面的Notch配體胞外區的結合。脊椎動物Notch配體分為Delta和Jagged 2個家族,人的Notch配體有Delta-1、-3、-4和Jagged-1、-2,人的Notch受體家族有4個成員(Notch1~4)。Notch信號途徑不僅影響著胚胎發育、血管發生、程序性細胞死亡和細胞增殖等多種生理過程[1-5],在腫瘤的侵襲、轉移、凋亡、增殖和血管生成等病理過程中也發揮著關鍵作用[6-7]。本研究通過構建shRNA-Notch1真核表達質粒以探究沉默Notch1基因對人乳腺癌MCF-7細胞凋亡的影響及其分子機制。
材 料 和 方 法
1材料
MCF-7細胞購自中國科學院細胞庫,大腸桿菌菌株DH5α由本校分子醫學實驗中心提供。胎牛血清和RPMI-1640培養基購自Gibco;pRS質粒載體購自Origene;質粒提取試劑盒、瓊脂糖凝膠DNA回收試劑盒購自北京天根公司;限制性核酸內切酶BamH I、Hind III、XhoI和T4 DNA連接酶購自TaKaRa;脂質體Lipofectamine 2000和Annexin V/PI檢測凋亡試劑盒購自Invitrogen; Notch1、Hes-1、p-JNK(Thr183/Tyr185)、p-p53(Ser15)、PUMA(p53-upregulated modulator of apoptosis)、NOXA、cleaved caspase-3和β-actin抗體購自Santa Cruz;Rhodamine123購自Sigma;Notch1特異性DNA干擾序列由大連寶生物公司合成。
2方法
2.1設計shRNA靶序列 利用NCBI數據庫檢索人Notch1 mRNA序列(GenBank Accession:NM_017617),根據shRNA設計原則,使用Invitrogen公司在線設計軟件針對Notch1編碼區(2 015~2 035)設計特異性靶序列,另設計一條由Notch1特異性靶序列隨機重排而成的序列作為陰性對照,經BLAST比對分析與人類基因編碼序列無同源性。shRNA設計模板采用BamH I+Sense+Loop+Antisense+終止序列+XhoI+HindIII(其中Loop: 5’-TTCAAGAGA-3’, 終止序列: TTTTTT),具體序列見表1,下劃線部分為靶序列,由大連寶生物公司合成。

表1 shRNA干擾序列
2.2構建pRS-shRNA表達質粒 將pRS空質粒于37 ℃用限制性核酸內切酶BamH I和HindIII酶切過夜,經1%瓊脂糖凝膠電泳后,采用瓊脂糖凝膠DNA回收試劑盒回收。將合成的shRNA模板單鏈進行退火處理形成雙鏈,然后與雙酶切后的線性化載體于16 ℃連接過夜。將連接產物轉化大腸桿菌DH5α感受態細胞,涂布于含ampicillin抗性的LB平板上,37 ℃培養過夜,挑取單克隆菌落,置于含有ampicillin抗性的LB培養基中,37 ℃、200 r/min過夜。收集菌液,采用質粒提取試劑盒提取質粒。
2.3重組質粒的鑒定 提取的質粒用XhoI做酶切鑒定。空質粒pRS的DNA序列中不存在XhoI的酶切位點,而在插入的shRNA片段中,我們加入了1個XhoI的酶切位點。若重組質粒構建成功,就能被XhoI酶切。對酶切鑒定正確的質粒送大連寶生物公司測序。鑒定正確的質粒分別命名為pRS-Notch1和pRS-non。
2.4細胞培養和轉染 用含10%胎牛血清的RPMI-1640培養基,于37 ℃、5% CO2培養箱中培養MCF-7細胞,每隔48 h更換1次細胞培養液。當細胞匯合度達80%以上時換為不含胎牛血清的RPMI-1640培養基培養過夜。將重組質粒pRS-Notch1和pRS-non分別轉染MCF-7細胞,轉染方法參照Lipofectamine 2000操作說明書進行,置于37 ℃、5% CO2培養箱中培養6 h后用PBS洗去轉染試劑,加入新鮮的培養基繼續培養24 h。MCF-7細胞轉染分組:(1)shControl組:轉染重組質粒pRS-non;(2)shNotch1組:轉染重組質粒pRS-Notch1。
2.5流式細胞術檢測細胞凋亡 制備各實驗組細胞懸液,用PBS洗滌細胞2次,加入結合緩沖液懸浮細胞,加入Annexin V-FITC輕輕混勻后于4 ℃避光孵育10 min。800 r/min離心5 min,棄上清,重懸細胞于結合緩沖液中,加入PI染色液輕輕混勻后于4 ℃避光孵育5 min,送流式細胞儀檢測。
2.6Western blotting檢測蛋白水平 裂解各組細胞提取總蛋白,用BCA法定量后,進行SDS-PAGE電泳并轉移至PVDF膜。封閉液(5%BSA/TBST)封閉1 h,加入Ⅰ抗(1∶1 000稀釋),4 ℃孵育過夜,TBST洗膜3次,加入Ⅱ抗(1∶1 000稀釋)室溫下孵育1 h,TBST洗膜3次,加入ECL進行發光反應,壓片、顯影、定影,觀察蛋白印記,運用ImageJ軟件測定各蛋白條帶灰度值。
2.7流式細胞術檢測線粒體膜電位 羅丹明123(rhodamine 123, Rh123)是一種可以通過細胞膜的選擇性染色活細胞線粒體的熒光染料,細胞內Rh123的平均熒光強度(mean fluorescence intensity, MFI)與線粒體跨膜電位呈正相關。制備各實驗組細胞懸液,用PBS洗滌細胞2次,重懸細胞于培養基中,加入Rh123(終濃度為5 mg/L),37 ℃孵育箱中放置30 min,再用細胞培養基洗滌細胞2次,送流式細胞儀檢測。
3統計學處理
用SPSS 16.0統計軟件進行分析。數據用均數±標準差(mean±SD)表示,兩組間均數比較采用t檢驗,以P<0.05為差異有統計學意義。
結 果
1pRS-shRNA重組質粒的鑒定
將重組質粒和空質粒分別用XhoI進行單酶切鑒定。空質粒pRS因其序列中無XhoI的酶切位點而不能被XhoI酶切;但在重組質粒pRS-Notch1和pRS-non的序列中插入了XhoI的酶切位點,可被XhoI酶切,酶切產生的線狀DNA因空間位阻較環狀質粒大導致電泳速度慢于環狀質粒。對酶切鑒定正確的質粒測序, 經過比對,2個重組質粒中含有目的shRNA片段,表明重組質粒構建成功,見圖1。

Figure 1. Single restriction endonuclease digestion of pRS-Notch1 and pRS-non. M: 1 kb DNA marker; 1~3: pRS, pRS-Notch1 and pRS-non without digestion,respectively; 4~6: pRS, pRS-Notch1 and pRS-non digested byXhoI,respectively.
圖1重組質粒pRS-Notch1和pRS-non的單酶切鑒定
2shRNA干擾對Notch1和Hes-1蛋白表達的影響
Western blotting結果顯示,shNotch1組Notch1/β-actin灰度比顯著低于shControl組(P<0.01)。Hes-1基因是Notch1的主要下游基因之一,當shNotch1組Notch1蛋白表達被抑制后,該組Hes-1蛋白表達較shControl組明顯下降(P<0.01),這表明Notch1信號途徑受到明顯抑制,見圖2。
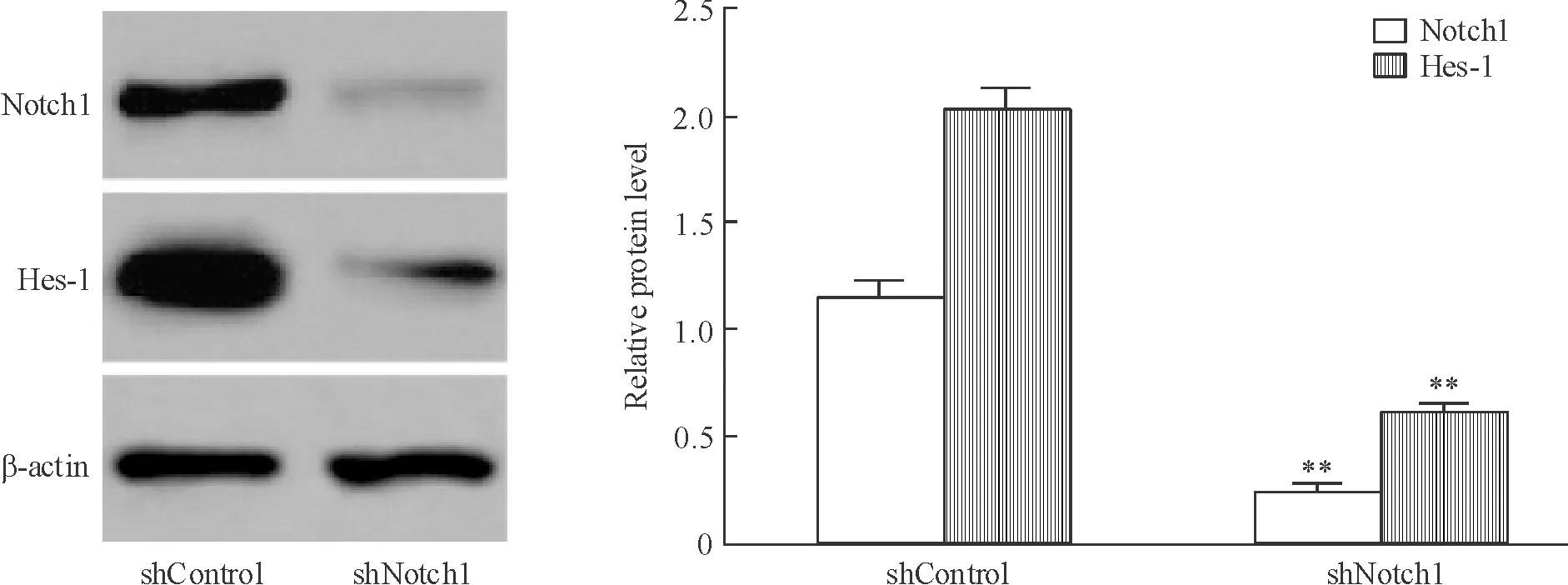
Figure 2. Expression of Notch1 and Hes-1 proteins in human breast cancer MCF-7 cells after pRS-non/pRS-Notch1 transfection. Mean±SD.n=3.**P<0.01vsshControl group.
圖2pRS-non/pRS-Notch1轉染MCF-7細胞后Notch1和Hes-1蛋白的表達
3沉默Notch1基因對細胞凋亡的影響
流式細胞術結果顯示,shNotch1組MCF-7細胞凋亡率為23.8%±1.7%,較shControl組(2.1%±0.5%)顯著升高(P<0.01),見圖3。
4沉默Notch1基因對JNK1和p53磷酸化的影響
shNotch1組p-JNK1/β-actin和p-p53/β-actin的灰度比分別為1.050 3±0.071 2和0.830 2±0.063 5,均顯著高于shControl組(P<0.05),見圖4。
5沉默Notch1基因對PUMA、NOXA和cleavedcaspase-3蛋白表達的影響
shNotch1組PUMA/β-actin、NOXA/β-actin和cleaved caspase-3/β-actin的灰度比分別為0.827 1±0.070 8、1.737 6±0.096 3和1.234 1±0.078 2,均顯著高于shControl組,(P<0.05),見圖5。

Figure 3. Effect ofNotch1 silencing on apoptosis of MCF-7 cells detected by flow cytometry. Mean±SD.n=3.**P<0.01vsshControl group.
圖3流式細胞術檢測沉默Notch1對MCF-7細胞凋亡的影響
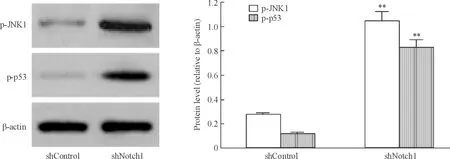
Figure 4. Effect ofNotch1 silencing on phosphorylations of JNK1 and p53 in MCF-7 cells. Mean±SD.n=3.**P<0.01vsshControl group.
圖4沉默Notch1對MCF-7細胞JNK1和p53磷酸化的影響
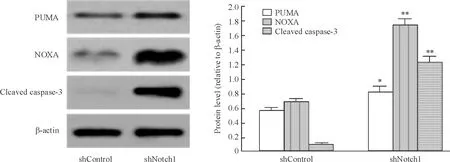
Figure 5. Effect ofNotch1 silencing on the protein levels of PUMA,NOXA and cleaved caspase-3 in MCF-7 cells. Mean±SD.n=3.*P<0.05,**P<0.01vsshControl group.
圖5沉默Notch1對MCF-7細胞PUMA、NOXA和cleavedcaspase-3蛋白水平的影響
6沉默Notch1基因對線粒體膜電位的影響
流式細胞術結果顯示,shControl組的FI為5 576±410,以此為對照,沉默Notch1基因后MCF-7細胞的FI降為4 332±338 (P<0.05),表明抑制Notch1蛋白表達可引起MCF-7細胞線粒體膜電位下降,見圖6。
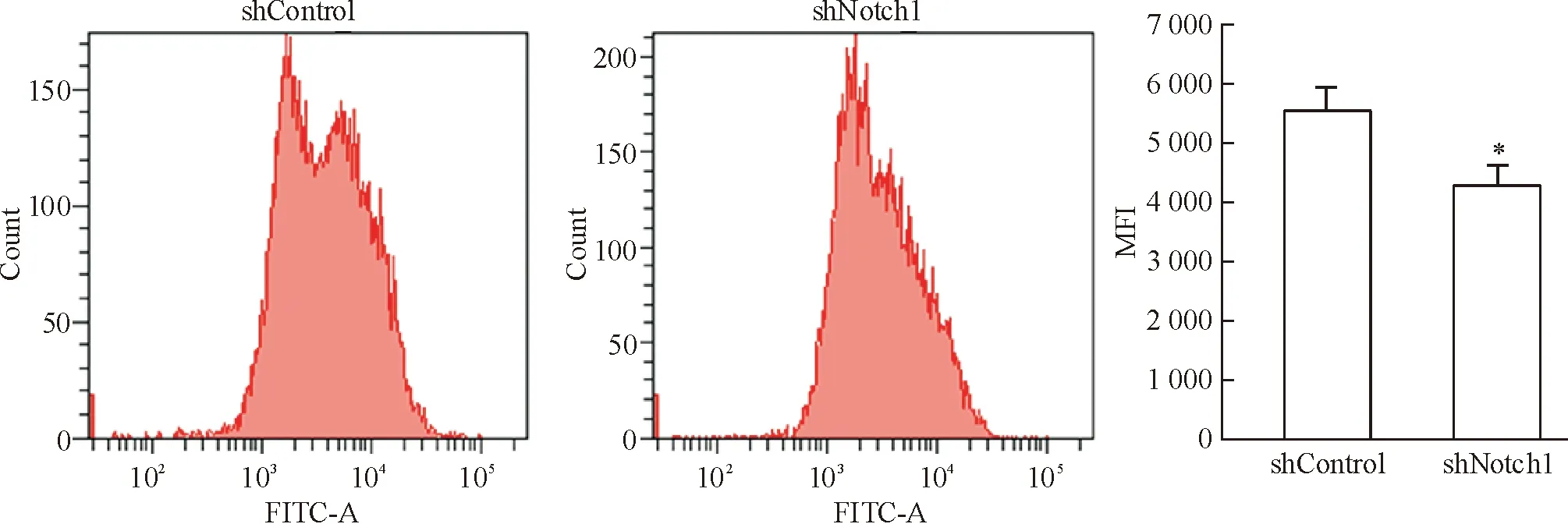
Figure 6. Effect ofNotch1 silencing on mitochondrial membrane potential of MCF-7 cells detected by flow cytometry. Mean±SD.n=3.*P<0.05vsshControl group.
圖6流式細胞術檢測沉默Notch1基因對MCF-7細胞線粒體膜電位的影響
討 論
Notch1單鏈前體(300 kD)首先在內質網合成,運輸至高爾基體后被Furin 樣轉化酶切割成含胞外區的N端片段(180 kD)與含跨膜區和胞內區的C端片段(120 kD),兩者通過非共價鍵結合為異源二聚體后被轉運到細胞膜。當Notch與相鄰細胞表面的配體結合后,Notch1相繼發生2次蛋白水解。先由ADAM10/Kuz或ADAM17/TACE酶切其C端片段的胞外部分產生Notch1胞外截短體(Notch extracellular truncation, NEXT),再由γ-促分泌酶復合體酶切NEXT釋放Notch1胞內段 (Notch intracellular domain,NICD),NICD進入細胞核與CSL [CBF1/Su(H)/LAG1]結合[8]。CSL蛋白是一種轉錄抑制因子,在哺乳動物中又被稱為C-啟動子結合因子1 (C-promoter binding factor 1, CBF1)或轉錄因子重組信號結合蛋白 Jκ (recombination signal binding protein-Jκ, RBP-Jκ)。沒有NICD存在時,CSL與Notch1靶基因啟動子結合并抑制其轉錄,當NICD與CSL結合并募集Mastermind組成轉錄激活復合體后,CSL由轉錄抑制因子轉變為轉錄激活因子,激活Notch1靶基因的轉錄,如Hes-1、Hey-1、CDK8和CCND1等[9]。
本研究針對Notch1的開放讀碼框設計了特異性的shRNA干擾序列,以此構建的重組質粒pRS-Notch1在轉染人乳腺癌MCF-7細胞后有效抑制了Notch1基因的表達。為進一步明確Notch1信號通路是否被有效抑制,我們檢測了Notch1信號通路的特異性靶基因Hes-1的表達水平,結果顯示,隨著Notch1蛋白表達的抑制,Hes-1蛋白的表達顯著降低,這表明重組質粒pRS-Notch1能夠有效沉默Notch1基因和抑制Notch1信號通路。
抑制Notch1信號通路可促進多種細胞發生凋亡,然而在不同的細胞環境中,分子機制不盡相同。運用γ-分泌酶抑制劑(γ-secretase inhibitor,GSI)阻斷Notch信號通路可下調骨髓瘤細胞Akt、Bcl-2和Bcl-xL蛋白的表達[10];通過siRNA沉默前列腺癌細胞Notch1基因可抑制Akt和轉錄因子FoxM1的表達[11],下調Bcl-2蛋白和上調Bax蛋白的表達[12]。在轉移性前列腺癌細胞中通過下調Notch1或Jagged1蛋白的表達抑制Notch1信號通路后,Akt和mTOR蛋白的表達均受到抑制,同時NF-κB信號通路及其下游蛋白MMP-9、VEGF和uPA的表達也均受到抑制[7]。
本研究發現,沉默乳腺癌MCF-7細胞Notch1基因可促進JNK1和p53的磷酸化。這可能是由于Notch1信號通路活化產生的NICD可與JNK1競爭結合JNK相互作用蛋白1(JNK-interacting protein 1, JIP1),從而干擾了JIP1-JNK1信號通路的活化[13],沉默Notch1基因后,解除了NICD對JIP1-JNK1信號通路的抑制,促進了JNK1的磷酸化。JNK1激活后可磷酸化p53使之活性增強[14]。在人肝癌HepG2細胞中,DNA損傷可引起p53蛋白磷酸化(Ser15), PUMA、NOXA、Bax和Bcl-2蛋白表達增多,繼而促使細胞色素C和Smac/DIABLO蛋白從線粒體膜間腔釋放至胞漿,激活caspase-9和caspase-3導致細胞凋亡[15]。
BH3-only蛋白PUMA和NOXA為p53的下游蛋白,可與Bcl-2蛋白家族中抗凋亡蛋白結合,促進Bax的活化,活化的Bax可插入線粒體外膜并寡聚成孔道,也可與線粒體外膜中的電壓依賴性陰離子通道 (voltage-dependent anion channel, VDAC)結合引起線粒體通透性轉變孔 (mitochondrial permeability transition pore, MPTP)的持續開放,進而引起線粒體外膜通透化 (mitochondrial outer membrane permeabilization, MOMP)和線粒體膜電位消散,釋放線粒體膜間腔中的促凋亡蛋白,激活半胱天冬酶(caspase),最終導致細胞凋亡[16-17]。本研究發現,在沉默乳腺癌MCF-7細胞Notch1基因后,隨著p53蛋白磷酸化水平的升高,PUMA和NOXA蛋白的表達明顯增強,進而導致線粒體膜電位降低和caspase-3活化。
[1] Fernandez-Valdivia R, Takeuchi H, Samarghandi A,et al. Regulation of mammalian Notch signaling and embryonic development by the proteinO-glucosyltransferase Rumi[J]. Development, 2011, 138(10):1925-1934.
[2] Gianni-Barrera R, Trani M, Reginato S,et al. To sprout or to split? VEGF, Notch and vascular morphogenesis[J]. Biochem Soc Trans, 2011, 39(6):1644-1648.
[3] Yalcin-Ozuysal ?, Fiche M, Guitierrez M,et al. Antagonistic roles of Notch and p63 in controlling mammary epithelial cell fates[J]. Cell Death Differ, 2010, 17(10):1600-1612.
[4] Monahan P, Rybak S, Raetzman LT. The Notch target geneHes1 regulates cell cycle inhibitor expression in the developing pituitary[J]. Endocrinology, 2009, 150(9):4386-4394.
[5] 郭 亞, 趙能江, 林 玲,等.Notch1基因對人膠質瘤U251細胞增殖和周期的影響[J].中國病理生理雜志, 2010, 26(6):1115-1119.
[6] Garcia A, Kandel JJ. Notch: a key regulator of tumor angiogenesis and metastasis[J]. Histol Histopathol, 2012, 27(2):151-156.
[7] Wang Z, Li Y, Banerjee S,et al. Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-κB signaling pathways[J]. J Cell Biochem, 2010, 109(4):726-736.
[8] Fortini ME. Notch signaling: the core pathway and its posttranslational regulation[J]. Dev Cell, 2009, 16(5): 633-647.
[9] Ranganathan P, Weaver KL, Capobianco AJ,et al. Notch signalling in solid tumours: a little bit of everything but not all the time[J]. Nat Rev Cancer, 2011, 11(5):338-351.
[10] Nefedova Y, Sullivan DM, Bolick SC,et al. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy[J]. Blood, 2008, 111(4):2220-2229.
[11] Wang Z, Li Y, Ahmad A,et al. Down-regulation of Notch-1 is associated with Akt and FoxM1 in inducing cell growth inhibition and apoptosis in prostate cancer cells[J]. J Cell Biochem, 2011, 112(1):78-88.
[12] Ye QF, Zhang YC, Peng XQ,et al. Silencing Notch-1 induces apoptosis and increases the chemosensitivity of prostate cancer cells to docetaxel through Bcl-2 and Bax[J]. Oncol Lett, 2012, 3(4): 879-884.
[13] Kim MY, Ann EJ, Mo JS,et al. JIP1 binding to RBP-Jκ mediates cross-talk between the Notch1 and JIP1-JNK signaling pathway[J]. Cell Death Differ, 2010, 17(11):1728-1738.
[14] Fan S, Qi M, Yu Y,et al. P53 activation plays a crucial role in silibinin induced ROS generation via PUMA and JNK[J]. Free Radic Res, 2012, 46(3):310-319.
[15] Yang G, Zhou X, Wang J,et al. MEHP-induced oxidative DNA damage and apoptosis in HepG2 cells correlates with p53-mediated mitochondria-dependent signaling pathway[J]. Food Chem Toxicol, 2012, 50(7):2424-2431.
[16] Chipuk JE, Moldoveanu T, Llambi F,et al. The BCL-2 family reunion[J]. Mol Cell, 2010, 37(3): 299-310.
[17] Whelan RS, Konstantinidis K, Wei AC,et al. Bax regulates primary necrosis through mitochondrial dynamics[J]. Proc Natl Acad Sci U S A, 2012, 109(17):6566-6571.
Notch-1genesilencingpromotesphosphorylationsofJNK1andp53inhumanbreastcancerMCF-7cells
YUAN Lei, CHEN Xu-dong, FAN Wen-juan, YANG Xu-guang, WANG Jian-guo
(LuoheMedicalCollege,Luohe462002,China.E-mail:wr0395@sina.com)
AIM: To investigate the effect ofNotch1 gene silencing on phosphorylations of JNK1 and p53 in human breast cancer MCF-7 cells.METHODSshRNA-Notch1 eukaryotic expression plasmid was constructed and transfected into MCF-7 cells. The expression of Notch1 and Hes-1 was observed by Western blotting after transfction. Apoptosis and mitochondrial membrane potential were detected by flow cytometry. Western blotting was also used to determine the protein levels of p-JNK1, p-p53, PUMA, NOXA and cleaved caspase-3 afterNotch1 silencing was performed in MCF-7 cells.RESULTSSilencing ofNotch1 significantly reduced the expression of Notch1 and Hes-1 in MCF-7 cells (P<0.01). In shNotch1 group, the number of apoptotic cells was much higher (P<0.01) and mitochondrial membrane potential was much lower (P<0.05) than those in shControl group. The protein levels of p-JNK1, p-p53, PUMA, NOXA and cleaved caspase-3 increased obviously after silencing ofNotch1 was performed in MCF-7 cells (P<0.05).CONCLUSIONNotch1 silencing induces apoptosis of human breast cancer MCF-7 cells through promoting phosphorylations of JNK1 and p53, and increasing the production of PUMA, NOXA and cleaved caspase-3.
Notch1 protein; Short hairpin RNA; JNK1 protein; p53 protein; MCF-7 cells
R737.9
A
10.3969/j.issn.1000- 4718.2013.06.010
1000- 4718(2013)06- 1014- 06
2012- 12- 31
2013- 04- 02
河南省基礎與前沿技術研究計劃項目(No.122300410277);漯河醫學高等專科學校科研基金資助項目(No.2010-S10)
△通訊作者 Tel: 0395-2112681; E-mail: wr0395@sina.com
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
