siRNA靶向干擾組蛋白H3表達對人鼻咽癌細胞增殖的抑制作用*
2013-10-24 01:44:28李彬彬黃國良董子明何志巍
中國病理生理雜志 2013年9期
李彬彬, 黃國良, 孔 霞, 李 蓉, 董子明, 何志巍,△
(廣東醫學院 1病理生理學教研室, 2廣東省醫學分子診斷重點實驗室,廣東 東莞 523808;3鄭州大學基礎醫學院病理生理學教研室,河南 鄭州 450001)
siRNA靶向干擾組蛋白H3表達對人鼻咽癌細胞增殖的抑制作用*
李彬彬1, 黃國良2, 孔 霞1, 李 蓉1, 董子明3, 何志巍1,2△
(廣東醫學院1病理生理學教研室,2廣東省醫學分子診斷重點實驗室,廣東 東莞 523808;3鄭州大學基礎醫學院病理生理學教研室,河南 鄭州 450001)
目的應用RNA干擾技術抑制人鼻咽癌細胞株CNE1中組蛋白H3的表達,觀察其對鼻咽癌細胞增殖的影響。方法構建針對組蛋白H3的小干擾RNA(siRNA)的真核表達載體,轉染CNE1細胞并篩選獲得穩定表達細胞株。實時熒光定量RT-PCR和Western blotting檢測細胞中組蛋白H3 mRNA和蛋白表達的改變;CCK-8法和平板克隆形成實驗觀察細胞增殖能力的改變;雙螢光素酶報告系統檢測細胞激活蛋白1(AP-1)轉錄活性的改變。結果與陰性對照組和空白對照組相比,siRNA-H3質粒轉染組的組蛋白H3 mRNA和蛋白表達均顯著下降,細胞生長速度明顯減慢,表皮生長因子誘導的細胞克隆形成能力和AP-1轉錄活性均受到明顯抑制。結論通過RNA干擾技術阻斷組蛋白H3的表達,可抑制CNE1細胞的生長、增殖,其機制可能與下調AP-1轉錄活性有關。組蛋白H3 可能是潛在的腫瘤治療新靶點。
鼻咽腫瘤; 組蛋白H3; RNA干擾; 激活蛋白1
表觀遺傳(epigenetics)是指DNA序列不發生變化但基因表達卻發生了可遺傳的改變[1]。通常DNA序列的改變是永久性的,而許多表觀遺傳的改變則是可逆的。表觀遺傳修飾主要包括以下4個方面:DNA甲基化、組蛋白修飾、染色質重構和非編碼RNA的調控。任何一個方面的異常都將影響染色體結構和基因表達,導致多種疾病及腫瘤的發生[2]。組蛋白修飾是細胞內基因轉錄調控的重要信息平臺,它通過整合上游分子通路,產生適宜的細胞核信號,如轉錄激活或轉錄抑制,從而調節基因的表達。近年來大量研究顯示,組蛋白H3異常修飾介導了腫瘤發生、演進的多個過程,是腫瘤表觀遺傳學的重要機制[2-3]。激活蛋白1 (activator protein-1, AP-1)是細胞內重要的核轉錄因子, 參與調控細胞增殖、分化和轉化等過程,促進腫瘤的發生、發展[4-5]。但對AP-1轉錄活性的調控機制目前尚未闡明。RNA干擾(RNA interference, RNAi)是一種選擇性沉默基因表達的有效工具,對此,我們構建了U6啟動子驅動的、組蛋白H3基因的小干擾RNA (small interfering RNA, siRNA)表達質粒,降低人鼻咽癌CNE1細胞中組蛋白H3的表達,并探討其對CNE1細胞增殖的影響和機制。
材 料 和 方 法
1材料和主要試劑
1.1菌株、細胞和質粒 大腸桿菌DH5α和人鼻咽癌CNE1細胞為本實驗室保存;mU6pro質粒由美國明尼蘇達大學惠贈;pcDNA3.1質粒由廣東醫學院張湘寧博士惠贈;pRTU14 AP-1 reporter vector 由德國Helmholtz Zentrum München研究中心惠贈。
1.2主要試劑 限制性內切酶BbsⅠ和XbaⅠ均購自NEB;T4 DNA連接酶購自大連TaKaRa;兔抗人組蛋白H3和H2A多抗購自Cell Signaling;紅外標記羊抗兔Ⅱ抗購自Rockland;JetPEI 轉染試劑購自Polyplus;G418和表皮生長因子(epidermal growth factor, EGF)購自Sigma-Aldrich;Cell Counting Kit-8 (CCK-8) 試劑盒購自日本同仁化學研究所;Dual-Luciferase Reporter Assay System 購自Promega。
2方法
2.1組蛋白H3 siRNA序列的設計和合成 在NCBI數據庫中查找人類組蛋白H3 mRNA的序列(NM_003537),其編碼序列(coding sequence,CDS)為1~411 bp。參考Choi等[6]的研究結果,選取16~34位(CAGACAGCTCGGAAATCCA)作為靶位點。按照mU6pro質粒的結構,設計包含BbsⅠ和XbaⅠ酶切殘端并能表達發夾結構RNA的2條寡核苷酸序列: 正義鏈5’-TTTGCAGACAGCTCGGAAATCCATTCAAGA-GATGGATTTCCGAGCTGTCTGTTTTTT-3’,反義鏈5’-CTAGAAAAAACAGACAGCTCGGAAATCCATCTCTT-GAATGGATTTCCGAGCTGTCTG-3’。同時,設計1對陰性對照序列,經BLAST同源比對,此序列不與任何人類基因序列同源。所有序列由上海生工生物工程技術服務有限公司合成。
2.2真核表達質粒mU6pro-siRNA-H3的構建 建立退火體系:50 μmol/L正反義鏈模板10 μL,5×Annealing Buffer 5 μL,加滅菌ddH2O 至50 μL。在PCR儀上進行退火反應:95 ℃ 2 min,每90 s下降1 ℃,降至25 ℃,-20 ℃保存。BbsⅠ和XbaⅠ雙酶切mU6pro質粒,純化后的線性化載體與退火產物連接,構建重組質粒mU6pro-siRNA-H3(si-H3)和陰性對照質粒mU6pro-siRNA-mock(si-mock)。轉化感受態細菌DH5α,用含氨芐青霉素的LB平板培養基培養并篩選轉化子,挑取單個菌落,在含100 mg/L氨芐青霉素的LB液體培養基中擴大培養。取培養好的菌液進行菌落PCR法鑒定,上游引物在U6啟動子區域設計:5’-ATATCCCTTGGAGAAAAGCCCTT-3’,而下游引物則是該質粒上M13R2的序列: 5’-CACAGGAAACAGCTATGACCAT-3’。陽性克隆送上海英駿生物技術有限公司測序。
2.3基因轉染及穩定表達細胞株篩選 CNE1細胞常規培養于含10%胎牛血清RPMI-1640培養基中,以5×105cells/well接種于6孔板中,培養至70%~80%融合,使用Polyplus 公司的JetPEI 轉染試劑進行si-H3和si-mock質粒的轉染。因mU6pro質粒缺乏抗性篩選標記,故與pcDNA3.1質粒按10∶1進行共轉染,具體操作按說明書進行。轉染后24 h,將細胞以1∶10的比例傳代至10 cm 培養板皿中,次日加入選擇性抗生素G418 200 mg/L進行加壓篩選,構建穩定干擾組蛋白H3的CNE1細胞株。
2.4實時熒光定量RT-PCR 檢測組蛋白H3 mRNA的表達 Trizol試劑提取細胞總RNA,按照Roche逆轉錄試劑盒說明合成cDNA第1鏈;以cDNA為模板,7500實時定量PCR儀進行實時定量PCR反應。組蛋白H3上游引物5’-GTTGCTGATTCGGAAGCTGC -3’,下游引物5’-GAAGCGAAGATCGGTCTTGAA-3’;GAPDH上游引物5’-CTCCTCCTGTTCGACAGTCAGC-3’,下游引物5’- CCCAATACGACCAAATCCGTT -3’。檢測各模板的閾值循環數(Ct),ΔΔCt=(Ct目的基因-CtGAPDH)實驗組-(Ct目的基因-CtGAPDH)對照組,以2-ΔΔCt表示實驗組目的基因的表達相對于對照組的變化倍數。
2.5Western blotting檢測轉染細胞組蛋白H3的表達 酸溶性蛋白抽提法提取組蛋白[7]:收集的細胞用PBS洗2次,加入細胞裂解液 (10 mmol/L HEPES,pH 7.9,1.5 mmol/L MgC12,10 mmol/L KCl,0.5 mmol/L DTT和1.5 mmol/L PMSF),冰浴1 h;4 ℃、8 000 r/min離心10 min,棄上清,沉淀用0.2 mol/L H2SO4重懸,冰浴30 min;收集上清液,加人5倍體積的冰丙酮,-20 ℃沉淀過夜;收集沉淀蛋白,用去離子水溶解沉淀,-80 ℃保存。
取等量樣品,進行15%SDS-PAGE,將凝膠中的蛋白轉移到硝酸纖維素膜上,5%脫脂奶粉封閉2 h;加入組蛋白H3抗體(1∶1 000),4 ℃孵育過夜;加入紅外標記的羊抗兔Ⅱ抗(1∶10 000),室溫孵育1 h;Odyssey近紅外雙色激光成像系統掃描成像,組蛋白H2A作為內參照,Quantity One軟件進行半定量分析。
2.6CCK-8法檢測細胞體外增殖能力 取對數生長期的細胞,以1 000 cells/well接種到96孔板,每組設5個平行孔并設不含細胞的空白對照,37 ℃連續培養5 d;每天于同一時間取出加入CCK-8試劑10 μL/well,繼續培養2 h,在多功能酶標儀上測定450 nm波長各孔吸光度(A450)。根據每組細胞A450值繪制生長曲線。
2.7平板克隆形成實驗檢測細胞克隆形成能力 取對數生長期的細胞,以300 cells/well接種于含EGF(0 μg/L或10 μg/L)培養基的6孔板中,每組設3個平行孔,靜置培養2周;用PBS清洗細胞2次,甲醇固定15 min后,0.4%結晶紫染色15 min,流水沖洗;將平皿倒置并疊加1張帶網格的透明膠片,用肉眼直接計數克隆。
2.8雙螢光素酶報告基因分析檢測AP-1轉錄活性 取對數生長期的細胞,以1.0×105cells/well接種到24孔板,每組設3個平行孔,次日共轉染AP-1-luc 報告質粒和pRL-TK 內參照質粒。轉染24 h后移去培養基,換用無血清培養基饑餓24 h后用30 μg/L EGF刺激3 h,加入100 μL細胞裂解液,室溫下輕緩晃動15 min。收集細胞裂解液,按Dual-Luciferase Reporter Assay System 說明書分別測定螢火蟲螢光素酶和海腎螢光素酶的活性,以兩者的比值反映AP-1轉錄活性。
3統計學處理
應用SPSS 17.0及GraphPad Prism軟件分析。計量資料采用均數±標準差(mean±SD)表示,組間均數比較采用單因素方差分析,以P<0.05為差異有統計學意義。
結 果
1重組質粒si-H3的PCR鑒定和測序結果
重組質粒si-H3及其陰性對照質粒si-mock分別轉化大腸桿菌DH5α,挑選氨芐青霉素抗性菌液進行陽性克隆菌液PCR鑒定,陽性克隆的PCR產物大小為385 bp。將陽性克隆菌液進行測序,結果與所設計的寡核苷酸序列一致, 表明si-H3和si-mock真核表達質粒構建成功。
2穩定干擾組蛋白H3CNE1細胞株的構建
將si-H3質粒、si-mock質粒與pcDNA3.1質粒共轉染CNE1細胞,采用G418加壓篩選獲得穩定干擾組蛋白H3的CNE1細胞株及陰性對照細胞株。采用實時熒光定量RT-PCR(圖1)和Western blotting(圖2)檢測組蛋白H3 mRNA和蛋白表達水平。與空白對照組(blank)相比,陰性對照組(si-mock)細胞組蛋白H3的mRNA和蛋白表達均無明顯差異。與空白對照及陰性對照組相比,轉染si-H3質粒的CNE1細胞組蛋白H3的mRNA出現明顯降低,約為陰性對照組的33.6%,差異有統計學意義(P<0.05)。與空白對照及陰性對照組相比,轉染si-H3質粒的CNE1細胞組蛋白H3的蛋白表達水平亦明顯下調,約為陰性對照組的38.8%,差異有統計學意義(P<0.05)。這表明已成功構建穩定干擾組蛋白H3表達的CNE1細胞株。
3siRNA-H3對CNE1細胞增殖的影響
在細胞接種后1~5 d,以CCK-8法檢測細胞增殖能力。與空白對照組相比,陰性對照組細胞的增殖能力無明顯改變;但與空白對照組和陰性對照組相比,轉染si-H3質粒的CNE1細胞在48 h后生長速度明顯減慢,細胞群體倍增時間延長,差異有統計學意義(P<0.01),見圖3。這表明siRNA下調組蛋白H3的表達可明顯抑制CNE1細胞的增殖。
4siRNA-H3對CNE1細胞克隆形成能力的影響
EGF作為腫瘤促進因子,如圖4所示,可以促進CNE1細胞的克隆形成能力,主要引起集落形成大小的增加。在無或有EGF(10 μg/L)刺激下,與空白對照組相比,陰性對照組細胞的克隆形成數目及集落形成大小均無明顯改變;但與空白對照組和陰性對照組比較,轉染si-H3質粒的CNE1細胞的集落形成數目及集落形成大小均明顯減少,差異有統計學意義(P<0.05),見圖4。表明在有或無EGF刺激下,下調組蛋白H3表達均可有效抑制CNE1細胞的克隆形成能力。

Figure 1. The mRNA level of histone H3 in stably transfected cells detected by real-time fluorescence quantitative RT-PCR. Mean±SD.n=3.*P<0.05vssi-mock.
圖1實時熒光定量RT-PCR檢測轉染細胞中組蛋白H3mRNA的表達
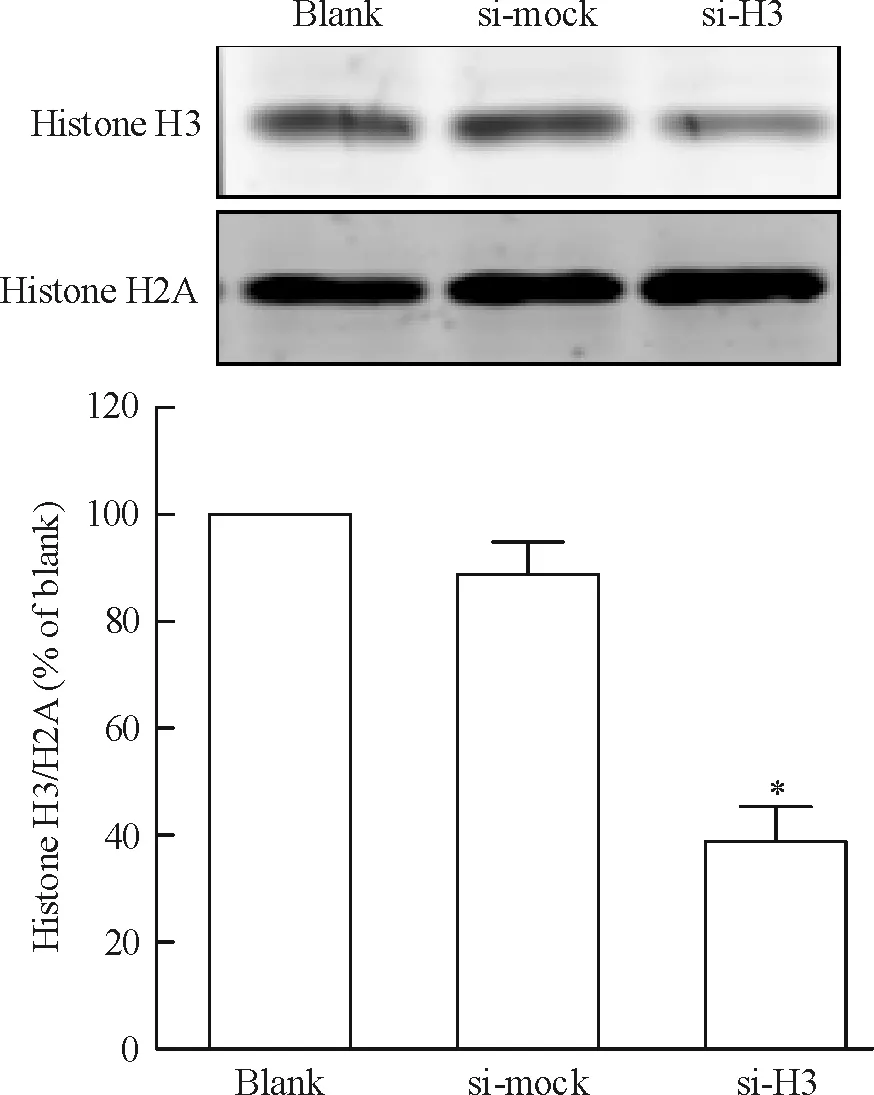
Figure 2. The expression of histone H3 protein in stably transfected cells detected by Western blotting. Mean±SD.n= 3.*P<0.05vssi-mock.
圖2Westernblotting檢測轉染細胞中組蛋白H3的表達
Figure 3. The effect of histone H3 knockdown on CNE1 cell proliferation. Mean±SD.n=3.*P<0.05.**P<0.01vssi-mock group.
圖3干擾組蛋白H3表達對CNE1細胞增殖的影響
Figure 4. The effect of histone H3 knockdown on colony formation of CNE1 cells. Mean±SD.n=3.*P<0.05,**P<0.01vssi-mock.
圖4干擾組蛋白H3表達對CNE1細胞克隆形成能力的影響
5siRNA-H3對CNE1細胞AP-1轉錄活性的影響
如圖5所示,EGF (30 μg/L) 可以明顯增強CNE1細胞轉錄因子AP-1的啟動子活性,約為對照組的2.82倍。在無或有EGF(30 μg/L)刺激下,與陰性對照組相比,陰性對照組細胞的AP-1的轉錄活性均無明顯改變;但與空白對照組和陰性對照組比較,轉染si-H3質粒的CNE1細胞轉錄因子AP-1啟動子活性明顯降低,與空白對照組相比分別下降了約52.8%和73.1%,差異有統計學意義(P<0.05)。表明在有或無EGF刺激下,下調組蛋白H3表達均可明顯抑制CNE1細胞的AP-1轉錄活性。

Figure 5. The effect of histone H3 knockdown on AP-1 transcriptional activation in CNE1 cells. Mean±SD.n=3.*P<0.05,**P<0.01vssi-mock.
圖5干擾組蛋白H3表達對CNE1細胞AP-1轉錄活性的影響
討 論
核小體是染色質的基本單位,核小體的核心由組蛋白H2A、H2B、H3 和H4各2個分子形成的八聚體和纏繞在上面的DNA分子構成。組蛋白在翻譯完成后,其N-末端氨基酸殘基會發生乙酰化、磷酸化、甲基化、泛素化、糖基化等共價修飾,從而提供一種識別的標志,稱為組蛋白密碼[8]。組蛋白的這些修飾幾乎都能影響染色質的結構和功能,進而改變組蛋白和DNA的相互作用,在基因轉錄調控中起關鍵作用。
大量研究表明[2-3],組蛋白修飾異常可引起相應染色體結構和基因轉錄水平的改變,影響細胞增殖、分化和凋亡,在腫瘤發生、發展中起重要作用。Feil等[9]報道,不利環境因素、飲食習慣等可以在不改變DNA序列的情況下,通過DNA甲基化和組蛋白共價修飾來擾亂相關基因的轉錄和表達,從而介導某些腫瘤的發生。近年來,組蛋白H3修飾異常與腫瘤發生的關系取得很大的進展。Tzao等[10]發現食管鱗癌患者中H3 K18乙酰化和H3 K27三甲基化水平與腫瘤分化程度呈正相關,H3 K27三甲基化水平低下的患者呈現較好的預后,提示H3 K27三甲基化水平可作為預測食管鱗癌患者預后的指標。H3 Ser10磷酸化是保證染色體濃縮和分離必不可少的因素之一。由AIM-1/Aurora B過表達引起的組蛋白H3 Ser10磷酸化水平的增高被發現在人類多種腫瘤中如結直腸癌、肝癌[11-12],可促進染色體的不穩定。我們在前面的研究中也發現[7],H3 Ser10磷酸化在低分化鼻咽癌組織中的表達明顯高于鼻咽炎癥組織和癌旁正常組織,并且與EB病毒潛伏性膜蛋白1(latent membrane protein 1,LMP1)的表達呈正相關關系。Nishikawa等[13]研究顯示,EB 病毒陽性而LMP1呈低表達的鼻咽癌細胞TWO3-EBV經乙烯基丁酸酯或去乙酰化酶抑制劑trichostatin A 處理后,出現LMP1 表達水平上升,同時,TWO3-EBV細胞中乙酰化組蛋白H3 和H4 的表達也持續上升,顯示組蛋白過乙酰化能上調EBV感染鼻咽癌細胞中的LMP1 的表達水平。
近年來研究發現組蛋白H3具有“癌基因”效應。Choi等[6]發現在小鼠JB6 C141細胞中過表達組蛋白H3可促進細胞的增殖和轉化,集落形成能力增強,而采用siRNA降低組蛋白H3的表達則可明顯抑制細胞的惡性轉化。AP-1屬于一類由即刻早期基因家族包括Jun、Fos和激活轉錄因子(activating transcription factor,ATF)組成的核轉錄因子。多種腫瘤刺激因子如EGF、佛波酯、紫外線以及氧化應激等均能誘導AP-1的激活。AP-1被發現在多種腫瘤組織中呈高表達,通過調控下游多個靶基因的轉錄活化在細胞增殖、轉化以及腫瘤細胞侵襲和轉移等過程中發揮作用[4-5]。近年來研究發現[6,14],組蛋白H3 Ser10磷酸化通過促進即刻早期反應基因c-jun、c-fos的轉錄激活進而調控AP-1的轉錄活性包括轉錄激活能力、DNA結合能力及穩定性,在EGF、佛波酯等促進的細胞惡性轉化中起重要作用。
本研究利用siRNA干擾CNE1細胞組蛋白H3基因,實時熒光定量RT-PCR和Western blotting結果顯示組蛋白H3表達在mRNA和蛋白水平均出現了明顯的降低,證明合成的siRNA能有效地沉默組蛋白H3基因,下調組蛋白H3的表達。在此基礎上,采用CCK-8 實驗和平板克隆形成實驗檢測CNE1細胞增殖能力的改變,結果顯示siRNA-H3沉默的CNE1細胞的增殖被顯著抑制,生長速度明顯減慢,其克隆形成數目和克隆形成大小均明顯減少。雙熒光素酶報告系統檢測顯示與細胞增殖相關的AP-1轉錄活性被明顯抑制。EGF是一強有力的促細胞分裂因子,它通過與EGF受體結合激活酪氨酸蛋白激酶活化引起的級聯反應促進細胞的增殖和轉化。本研究也發現,EGF可明顯促進CNE1細胞的增殖,并可增強AP-1的轉錄活性。但下調組蛋白H3的表達可顯著抑制EGF促進的細胞克隆形成能力及AP-1的轉錄活性。這些結果提示組蛋白H3作為正調控因子介導CNE1細胞的增殖和轉化,組蛋白H3對轉錄因子AP-1活性的調控可能是組蛋白H3介導細胞惡性轉化的重要機制。組蛋白H3有望成為腫瘤化療和基因治療的重要靶點。
[1] Esteller M. The necessity of a human epigenome project [J]. Carcinogenesis, 2006, 27(6):1121-1125.
[2] Jones PA, Baylin SB. The epigenomics of cancer [J]. Cell, 2007, 128(4):683-692.
[3] Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps [J]. Nat Rev Genet, 2007, 8(4):286-298.
[4] Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation [J]. Biochim Biophys Acta, 1991, 1072(2-3):129-157.
[5] 周長春, 劉芝華, 齊 軍,等. AP-1 和腫瘤的關系研究進展[J]. 世界華人消化雜志, 2006, 14(1):1-5.
[6] Choi HS, Choi BY, Cho YY, et al. Phosphorylation of histone H3 at serine 10 is indispensable for neoplastic cell transformation [J]. Cancer Res, 2005, 65(13): 5818-5827.
[7] Li B, Huang G, Zhang X, et al. Increased phosphorylation of histone H3 at serine 10 is involved in Epstein-Barr virus latent membrane protein-1-induced carcinogenesis of nasopharyngeal carcinoma [J]. BMC Cancer, 2013, 13:124.
[8] Jenuwein T, Allis CD. Translating the histone code [J]. Science, 2001, 293(5532): 1074-1080.
[9] Feil R. Environmental and nutritional effects on the epigenetic regulation of genes [J]. Mutat Res, 2006, 600(1-2):46-57.
[10] Tzao C, Tung HJ, Jin JS, et al. Prognostic significance of global histone modifications in resected squamous cell carcinoma of the esophagus [J]. Mod Pathol, 2009, 22(2):252-260.
[11] Sistayanarain A, Tsuneyama K, Zheng H, et al. Expression of Aurora-B kinase and phosphorylated histone H3 in hepatocellular carcinoma [J]. Anticancer Res, 2006, 26(5A): 3585-3593.
[12] Ota T, Suto S, Katayama H,et al. Increased mitotic phosphorylation of histone H3 attributable to AIM-1/Aurora-B overexpression contributes to chromosome number instability [J]. Cancer Res, 2002, 62(18):5168-5177.
[13] Nishikawa J, Kis LL, Liu A, et al. Upregulation of LMP1 expression by histone deacetylase inhibitors in an EBV carrying NPC cell line [J]. Virus Genes, 2004, 28(1):121-128.
[14] Clayton AL, Rose S, Barratt MJ, et al. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation [J].EMBO J, 2000, 19(14):3714-3726.
SmallinterferingRNA-mediatedsilencingofhistoneH3inhibitsproliferationofhumannasopharyngealcarcinomacells
LI Bin-bin1, HUANG Guo-liang2, KONG Xia1, LI Rong1, DONG Zi-ming3, HE Zhi-wei1,2
(1DepartmentofPathophysiology,2GuangdongProvincialKeyLaboratoryofMedicalMolecularDiagnostics,GuangdongMedicalCollege,Dongguan523808,China;3DepartmentofPathophysiology,BasicMedicalCollegeofZhengzhouUniversity,Zhengzhou450001,China.E-mail:zhiweihe688@yahoo.com)
AIM: To study the effect of histone H3 down-regulation by RNA interference on the proliferation of human nasopharyngeal carcinoma CNE1 cells.METHODSThe small interfering RNA (siRNA) vector targeting histone H3 was constructed and transfected into CNE1 cells, and then stably transfected CNE1 cells were established. The expression of histone H3 mRNA and protein was measured by real-time fluorescence quantitative RT-PCR and Western blotting, respectively. The proliferation ability of CNE1 cells was evaluated by CCK-8 and colony-forming assays. The transcriptional activity of activator protein-1 (AP-1) was examined by dual-luciferase reporter gene assay.RESULTSCompared with negative control and blank control groups, histone H3 mRNA and protein expression was markedly decreased in siRNA-H3 stably transfected CNE1 cells. Knockdown of histone H3 caused a significant inhibition of cell proliferation. Furthermore, the colony formation and AP-1 transcriptional activity promoted by epidermal growth factor were obviously suppressed.CONCLUSIONKnockdown of histone H3 could significantly inhibit the proliferation of CNE1 cells by down-regulating the AP-1 transcriptional activity, and therefore histone H3 might serve as a therapeutic target in cancer treatment.
Nasopharyngeal neoplasms; Histone H3; RNA interference; Activator protein-1
R739.6
A
1000- 4718(2013)09- 1625- 06
2013- 06- 07
2013- 07- 10
國家自然科學基金資助項目(No. 81071638);廣東省醫學科學技術研究基金資助項目(No. A2011424)
△通迅作者 Tel: 0769-22896324; E-mail: zhiweihe688@yahoo.com
10.3969/j.issn.1000- 4718.2013.09.015
