大黃素-鎂(Ⅱ)配合物的合成、表征及抗氧化活性研究
2013-11-01 03:18:10潘曉麗謝運飛章從恩董小萍
中成藥 2013年12期
關鍵詞:中藥
潘曉麗,向 暉,謝運飛,章從恩,董小萍
(成都中醫藥大學藥學院,中藥材標準化教育部重點實驗室,四川成都 611137)
自Rosenberg[1]發現順鉑的抗癌活性以來,無機成分中金屬元素的藥理活性日益受到人們的關注,基于金屬配合物的藥物研究得到了迅速發展[2]。許多事實證明,中藥中無機元素具有重要的藥理作用及活性,對于發揮中藥的療效有著十分重要的作用,中藥有效成分往往不單純是有機成分,而多是由有機成分和金屬元素組成的配合物[3]。金屬離子與中藥化學成分形成配合物后往往改變或增強其活性,例如抗菌、抗腫瘤、抗氧化等[4]。將中藥有效成分與金屬離子進行配合發揮兩者的協同作用,是提高中藥化學成分活性的有效途徑[5-6]。
大黃素(1,3,8-三羥基-6-甲基蒽醌)由于其結構中有相鄰的羰基和羥基,能與金屬離子形成配合物。唐睿等[7]采用ICP-AES法測定了大黃中5種微量元素(鎂、鐵、錳、鋅、銅)的量,結果表明大黃生藥中鎂的量最高。本實驗通過合成大黃素-鎂(Ⅱ)金屬配合物,研究其可能的配位結構及抗氧化活性,可以從一個嶄新的角度探索中藥新藥的開發方法,具有很大的研究意義。
1 試劑與儀器
核磁共振氫譜采用BRUKER AM—400型超導核磁共振儀(DMSO-d6作溶劑測定);紅外光譜采用BRUKER Tensor-27型傅立葉變換中紅外光譜儀測定;UV—1700型紫外分光光度計;PH計采用PHB—8型PH計。
大黃素(成都康邦生物生物科技有限公司,純度大于98.5%);二苯代苦味肼基自由基(DPPH·,美國Sigma公司),羥甲基氨基甲烷(tris,美國Sigma公司)、N-甲基吩嗪甲基硫酸鹽(PMS,美國Sigma公司),還原性輔酶I二鈉鹽(NADHNa2,美國Sigma公司),氯化硝基四氮唑藍(NBT,美國Sigma公司),C4H6O4Mg·4H2O,無水乙醇,無水甲醇等試劑為國產分析純,實驗用水為離子交換蒸餾水。
2 配合物的合成[8]
稱取1 mmoL大黃素(270 mg)溶于60 mL無水乙醇,40℃電磁攪拌,待大部分配體溶解后得黃色澄清溶液,加入0.5 mmoL(107.23 mg)C4H6O4Mg·4H2O(醋酸鎂)的15 mL無水乙醇溶液,逐滴加入氨水的乙醇溶液(1∶1,V∶V),調節反應溶液的pH值至9.5,溶液很快有橙紅色沉淀生成,繼續以反應溫度為40℃攪拌反應12 h,將溶液靜置過夜真空抽濾,依次用無水乙醇及乙醚分別將沉淀洗滌數次后,真空干燥72 h,得粉末狀固體產物。
3 配合物的EDTA滴定
稱取大黃素-鎂配合物約60 mg(記為m),在900℃馬弗爐中灼燒8 h至恒重得氧化鎂粉末,以稀硫酸溶解,加入pH=10的氨-氯化銨緩沖液5 mL,鉻黑T指示劑0.02 g,加入1∶1氨水數滴至產生白色氫氧化鎂沉淀,加水50 mL。以標定后的EDTA(Cedta)滴定,記下消耗的EDTA體積數(Vedta)。同時做空白實驗,其中金屬鎂含量計算公式為:
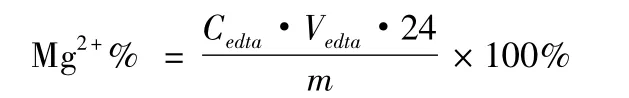
4 抗氧化活性實驗
4.1 配合物對DPPH·自由基的清除作用 取1 mL試樣與3 mL的8 mg/L DPPH無水甲醇溶液混均勻,暗室條件下反應30 min,于517 nm[9]處測定吸光度(A樣)。以1 mL甲醇代替試樣按照上述步驟進行反應,測定空白吸光度(A0)。
4.2 配合物對O2-·自由基的清除作用 由NADH/PMS體系產生[10],本實驗對其略作修改。反應體系為Tris-HCl溶液(0.05 mol/L,pH=8),其中含有 10μmol/L PMS,50μmol/L NADHNa2,25μmol/L NBT,以及不同質量濃度的1 mL試樣(分別取0.4 mg/L,0.2 mg/L,0.1 mg/L,0.05 mg/L,0.025 mg/L)。室溫反應5 min后,在386 nm波長處測定吸光值(A樣),1 mL蒸餾水代替試樣按照上述步驟進行反應,測定空白吸光度(A0)。
4.3 配合物對·OH自由基的清除作用 采用Fe2+-H2O2-亞甲藍體系[11],在5 mL量瓶中依次加入1 mLTris-HCl緩沖溶液(0.05 mol/L,pH=8),1.5 mL亞甲藍(0.02 g/L),0.5 mL FeSO4溶液(2 mmol/L),0.5 mL的0.5%H2O2溶液,以及不同質量濃度的1 mL試樣(分別取0.4 mg/L,0.2 mg/L,0.1 mg/L,0.05 mg/L,0.025 mg/L),蒸餾水稀釋至刻度,40℃水浴中加熱反應60 min,1 mL蒸餾水代替試樣按照上述步驟進行反應,測定空白吸光度(A0)。
以上自由基清除率計算公式為:清除率=[(A0-A樣)/A0]×100%
5 結果與討論
5.1 配合物的紫外光譜 將大黃素和大黃素-鎂(Ⅱ)配合物配成質量濃度為2.0×10-5mol/L的無水甲醇溶液,在200~500 nm范圍內進行UV-vis掃描分別得吸收光譜(見表1)。比較大黃素和大黃素-鎂(Ⅱ)配合物紫外吸收光譜可知,大黃素在219、253、289 nm處出現三個比較強吸收帶,是苯環共軛體系、醌樣結構譜帶,大黃素-鎂在223、264、293 nm處出現吸收帶,吸收峰位置發生一定程度移位,吸收強度減弱;大黃素在438 nm出現醌羰基吸收帶,而大黃素-鎂(Ⅱ)配合物在461 nm處出現吸收峰,比大黃素吸收帶紅移。峰位移動的原因可能是:大黃素與金屬離子形成配合物后,整個分子中電子的離域程度增大,致使電子躍遷時需要的能量降低,使吸收峰發生紅移。

表1 大黃素及大黃素-鎂(Ⅱ)的UV(nm)主要數據
5.2 配合物的紅外光譜 大黃素與大黃素-鎂(Ⅱ)配合物的紅外光譜主要特征峰(見表2)。配合物中原配體的特征峰1871 cm-1消失,同時配體在1624 cm-1(ν==C O)處的強吸收移至配合物的1601 cm(ν==C O),說明羰基氧與鎂配位;配體中酚羥基的伸縮振動的吸收峰3475 cm-1在配合物中顯著減弱,說明酚羥基與鎂配位。同時在592 nm-1出現了系Mg—O伸縮振動的吸收峰[12],進一步說明配合物的形成。
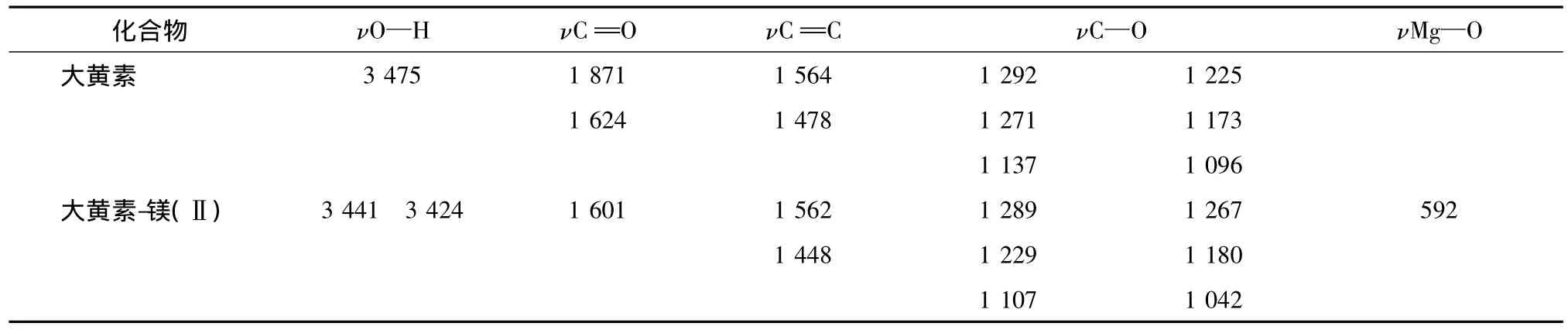
表2 大黃素及大黃素-鎂(Ⅱ)的IR(cm-1)主要數據
5.3 配合物的核磁共振氫譜 大黃素-鎂配合物的1H NMR(見表3)。δ 12.0~14(m,2H)出現一極弱多重峰,δ5.76~7.72(m,8H)出現一多重寬峰。考慮到大黃素的1,8位酚羥基性質相似,均可與9位羰基氧形成分子內氫鍵,由于3位羥基的給電子共軛效應及6位甲基的給電子誘導效應影響差異,可推測8位羥基易于解離。因此推測,配體分子中的8位酚羥基失去質子并通過9位氧原子與金屬離子形成配合物。
5.4 配合物的EDTA滴定結果 配合物中鎂離子測定結果為4.26%,理論值為4.27%。制得的大黃素-鎂配合物為兩分子大黃素與一分子鎂離子結合形成,實測值與理論值基本符合。結合上述分析結果,認為大黃素-鎂(Ⅱ)配合物的可能結構見結構圖1。

表3 大黃素及大黃素-鎂(Ⅱ)的1H NMR的數據(DMSO)
5.5 配合物的抗氧化活性
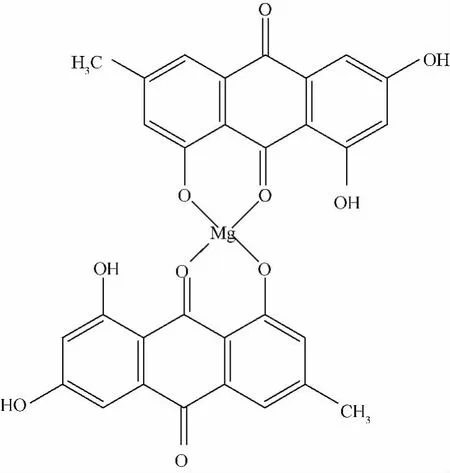
圖1 大黃素-鎂Ⅱ配合物結構圖
5.5.1 配合物對DPPH·自由基的清除作用 配合物和配體清除DPPH·自由基的結果見圖2,由圖可知,大黃素和大黃素-鎂均具有一定抗DPPH·自由基活性,且配合物的活性明顯比相同量的配體大黃素高,但抑制率不高,分析原因有可能是實驗在甲醇中進行,大黃素-鎂配合物在甲醇中比較穩定,不易形成穩定的自由基中間體。
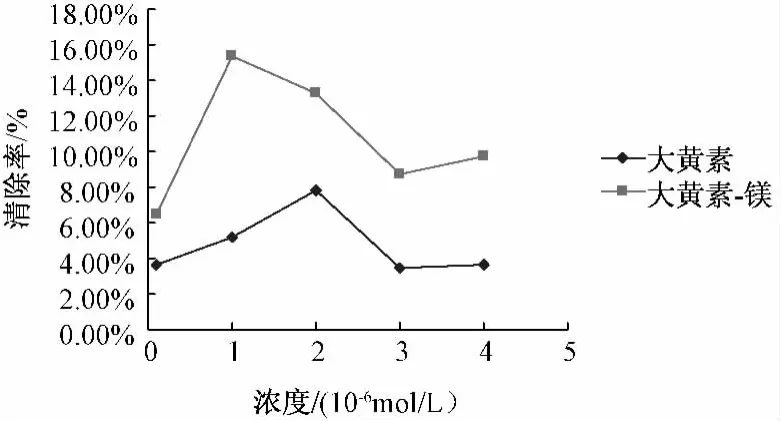
圖2 大黃素-Mg配合物及配體清除DPPH自由基活性
5.5.3 配合物對·OH自由基的清除作用 配合物和配體清除·OH自由基的結果見圖4,由圖可知,鎂配合物和配體大黃素均具有抗·OH自由基活性,相同濃度的鎂配合物抗·OH自由基活性較高,明顯高于配體大黃素,表明配體大黃素和鎂離子產生了協同作用,提高了其抗·OH的生物活性。
6 結論
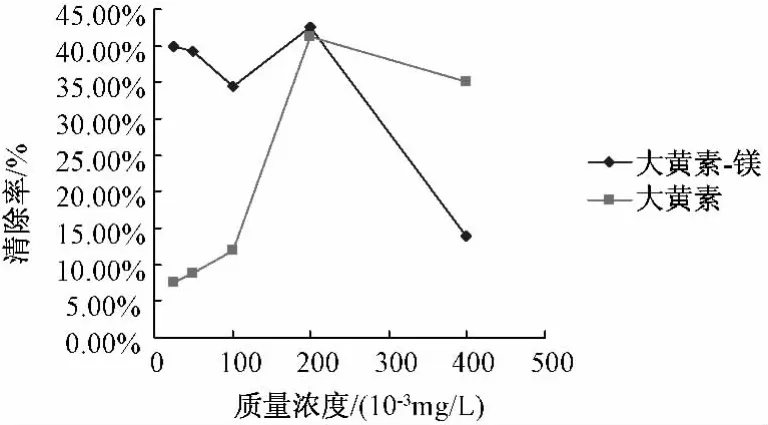
圖3 大黃素-Mg配合物及配體清除·自由基活性
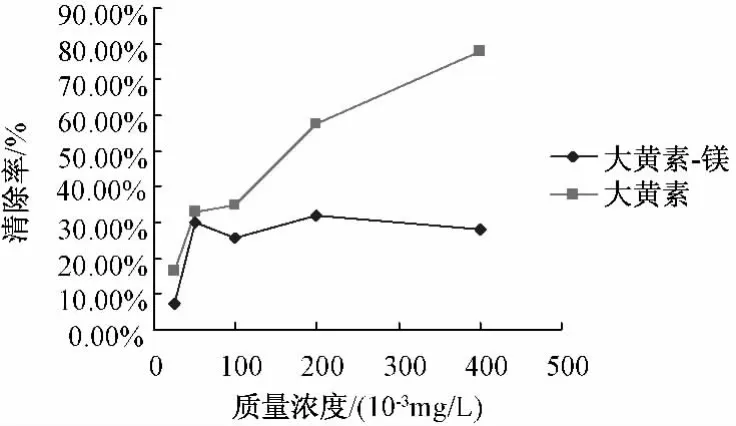
圖4 大黃素-Mg配合物及配體清除·OH自由基活性
本實驗以大黃素為配體合成了大黃素-Mg配合物,利用核磁共振氫譜,紫外光譜,紅外光譜,配位滴定等方法對其結構進行了表征,確定了配合物的可能組成。本實驗對比了大黃素與大黃素-Mg配合物對超氧陰離子自由基、羥基自由基、DPPH自由基等三種自由基的清除率,結果證明,大黃素-Mg配合物的抗氧化活性相對于配體大黃素有顯著提高,表明有可能是配體與金屬離子之間存在協同抗氧化作用。但由于配合物一分子含有兩分子大黃素配體,增加了酚羥基的數目,有實驗證明酚類物質的抗氧化性很多時候與酚羥基的數目及位置有關[13-15],故具體的構效關系還有待于進一步研究。
[1]Rosenberg B,Vancamp L,Trosko J E,et al.Platinum componds:a new class of potent antitumour agengts[J].Nature,1969,222(5191):385-386
[2]Orvig C,Abrams M J.Medicinal inorgantic chemistry:Introduction[J].Chem Rev,1999,99(9):2201-2204.
[3]朱旭祥,茅涵斌.中藥研究前沿一中藥配位化學[J].中草藥,1997,28(6):373-375.
[4]陳振鋒,彭 艷,譚明雄,等.基于中藥活性成分的金屬基抗腫瘤藥物前期研究[J].化學進展,2009,21(5):929-933
[5]譚明雄,陳振鋒,羅旭健,等.天然藥物有效成分的金屬配合物研究進展[J].林產化學與工業,2008,28(6):93-99
[6]鐘地長,張淑鳳,陳振鋒,等.天然產物黃酮類化合物的提取純化及其金屬配合物的研究進展[J].化學世界,2006(9):561-573
[7]唐 睿,溫金蓮,嚴志紅.ICP-AES法測定中藥大黃中五種微量元素[J].廣東微量元素科學.2005,12(12):38-40.
[8]趙 芳,梁 慧,程 慧,等.大黃酸金屬配合物的合成、表征及抗氧化活性研究[J].化學學報,2011,69(8):925-930.
[9]付萬平,張豫東,馬旭元,等.蘆薈大黃素鋅配合物的抗氧化活性[J].光譜實驗室,2012,29(2):1155-1157.
[10]Sun C,Wang J W,Lei F,et al.Free radical scavenging and antioxidant activities of EPS2,an exopolysaccha-ride produced by amarine filam en tous fungusK eissleriella sp.YS 4108[J].Life Sci,2004,75(9):1063-1073.
[11]李 芳,鄭懷禮.黃酮配合物抗自由基活性的亞甲基藍光測定體系的研究[J].光譜學與光譜分析學,2006,26(12):2294.
[12]中本一雄.無機和配位化合物的紅外和拉曼光譜[M].黃德如,譯.北京:科學出版社,2001.138.
[13]Rice-Evans C A,Miller N J,Bolwell G P,et al.The relative antioxidant activities of plant-derived polyphenolic flavonoids[J].Free Radic Res,1995,22(4):375-383.
[14]Husain S R,Cillard J,Cillard P.Hydroxyl Radical Scavenging Activity of Fla-vonoids[J].Phytochemistry,1987,26(9):2487-2491.
[15]Pokorny ,J Major factors affecting the antioxidant of lipids.in atuoxidation of unstaturated lipids(Chan H W S,ed)[M].London:Academic Press,1987:141-206.
猜你喜歡
中老年保健(2021年5期)2021-12-02 15:48:21
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
中國現代中藥(2020年10期)2020-12-16 08:53:18
金橋(2020年7期)2020-08-13 03:07:00
基層中醫藥(2020年12期)2020-07-22 06:34:38
中國現代中藥(2020年4期)2020-06-10 09:56:34
基層中醫藥(2018年6期)2018-08-29 01:20:20
長春中醫藥大學學報(2017年1期)2017-04-16 05:56:49
肝博士(2015年2期)2015-02-27 10:49:49
