α 干擾素增強(qiáng)骨肉瘤細(xì)胞對(duì)依托泊苷敏感性的實(shí)驗(yàn)研究
2014-01-18 03:09:24黃秀芳原向偉
中國醫(yī)藥導(dǎo)報(bào) 2014年7期
黃秀芳 原向偉
1.廣東省江門市中心醫(yī)院病理科,廣東江門 529030;2.廣東省江門市中心醫(yī)院骨科,廣東江門 529030
骨肉瘤高發(fā)于青少年人群,居于人類骨骼系統(tǒng)惡性腫瘤發(fā)病率第1 位,其惡性程度高,發(fā)展快,許多患者在發(fā)現(xiàn)時(shí)已存在肺轉(zhuǎn)移,因此療效不佳。 目前主要治療方法包括新輔助化療+外科手術(shù)包括保肢或截肢手術(shù),但能否手術(shù)及保證手術(shù)效果的重要前提是化療有效。臨床上相當(dāng)患者因?yàn)榛熌退幓蚨靖弊饔么蟮葐栴},引起化療失敗,導(dǎo)致患者無法行保肢手術(shù)從而截肢,甚至無法手術(shù)而導(dǎo)致死亡[1]。 因此,提高化療療效對(duì)于骨肉瘤的治療來講具有重大意義。IFN-α 屬于Ⅰ型干擾素,具有調(diào)節(jié)免疫、抗病毒、抗腫瘤等多種生物學(xué)作用[2],IFN-α 可直接抑制耐藥骨肉瘤細(xì)胞的生長[3],然而其對(duì)骨肉瘤化療敏感性的影響目前尚不清楚。因此筆者聯(lián)合使用IFN-α 與依托泊苷(Etoposide)處理人骨肉瘤U2OS 和MG63 細(xì)胞,探討其對(duì)兩種細(xì)胞生長抑制和凋亡的影響,并研究其分子機(jī)制,以期為提高骨肉瘤對(duì)化療藥物敏感性提供依據(jù)。
1 材料與方法
1.1 細(xì)胞培養(yǎng)
人骨肉瘤細(xì)胞U2OS(p53 基因野生型)和MG63(p53 基因突變型)購自中山大學(xué)病理教研室,培養(yǎng)液為DMEM(GIBCO 產(chǎn)品),其中含10%胎牛血清(GIBCO 產(chǎn)品)、100 U/mL 青霉素和100 μg/mL 鏈霉素,培養(yǎng)于37℃的5%CO2培養(yǎng)箱(Forma,美國)。
1.2 藥物處理
IFN-α 購自Peprotech 公司, 批號(hào)300-02A,Etoposide 購自Sigma 公司, 批號(hào)E1383, 稀釋分裝凍存-20℃, 使用時(shí)采用DMEM 培養(yǎng)液稀釋成工作濃度,處理分為四組:對(duì)照組(加入與等量于其余組的DMEM 培養(yǎng)液)、IFN-α (加入含工作濃度藥物的DMEM 培養(yǎng)液使終濃度為5000 U/mL) 組,Etoposide(終濃 度 在U2OS 細(xì) 胞 為2.5 μg/mL、MG63 細(xì) 胞 為0.625 μg/mL)組和IFN-α+Etoposide(終濃度同上)組。
1.3 流式細(xì)胞術(shù)檢測
細(xì)胞經(jīng)處理后, 消化離心, 收集細(xì)胞;PBS 洗兩次,離心;70%乙醇固定過夜;細(xì)胞經(jīng)10 μmol/L 的碘化丙啶(PI)染色5 min,4℃避光30 min;上FCM 流式細(xì)胞儀(BD,美國)用488 nm 激發(fā)光檢測細(xì)胞DNA含量,LYSIS 軟件分析凋亡率。
1.4 DNA Ladder 檢測
細(xì)胞經(jīng)各處理后,消化離心,收集細(xì)胞;PBS 洗兩次;加入0.8 mL 的DNAzol(Invitrogen 產(chǎn)品,批號(hào):10503-027),室溫放置數(shù)分鐘;4℃離心15 min;將上清移入新EP 管, 加入0.4 mL 的無水乙醇;4℃離心10 min;棄上清,加入75%乙醇洗2 次,加入適量TE 溶解基因組DNA,在含有EB 的1.8%瓊脂糖凝膠電泳(水平電泳槽為北京六一廠產(chǎn)品),紫外線激發(fā),凝膠成像分析儀(Bio-Rad,美國)采集DNA 梯形條帶。
1.5 Western blot
收集細(xì)胞,PBS 清洗離心,加入蛋白裂解液,定量蛋白(BCA 法),SDS-PAGE 電泳(垂直電泳儀為美國Bio-Rad 產(chǎn)品),然后電轉(zhuǎn)至PVDF 膜(電轉(zhuǎn)儀為美國Bio-Rad 產(chǎn)品),將膜封閉于含5%脫脂奶粉的TBST,4℃過夜; 鼠抗人p53、MDM2、Bax、Bcl-2、GAPDH 單克隆抗體和兔抗人PARP 多克隆抗體均為SantaCruz公司產(chǎn)品,稀釋后室溫封膜3 h 或4℃過夜,二抗稀釋后室溫封膜1 h 或4℃過夜, 暗室ECL 化學(xué)發(fā)光,膠片顯影,定影。
1.6 統(tǒng)計(jì)學(xué)方法
采用SPSS 14.0 軟件進(jìn)行統(tǒng)計(jì)分析, 計(jì)數(shù)資料以率表示,組間比較采用q 檢驗(yàn)。 以P < 0.05 為差異有統(tǒng)計(jì)學(xué)意義。
2 結(jié)果
2.1 IFN-α 在U2OS 和MG63 細(xì)胞對(duì)Etoposide 引起凋亡的影響
U2OS 和MG63 細(xì)胞經(jīng)IFN-α (5000 U/mL)和etoposide(U2OS 細(xì) 胞 為2.5 μg/mL、MG6 細(xì) 胞 為0.625 μg/mL)單獨(dú)或聯(lián)合處理72 h 后進(jìn)行流式細(xì)胞術(shù)檢測凋亡率,流式細(xì)胞術(shù)結(jié)果顯示,U20S 細(xì)胞在對(duì)照組、IFN-α 組、Etoposide 組和IFN-α+Etoposide 的細(xì)胞凋亡率分別為:(5.1±2.3)%、(5.7±2.7)%、(15.3±3.8)%和(45.5±5.2)%(圖1A),結(jié)果表明IFN-α 單獨(dú)并不誘導(dǎo)明顯的U2OS 細(xì)胞凋亡,卻明顯增強(qiáng)了Etoposide 誘導(dǎo)的U2OS 細(xì)胞凋亡,Etoposide 組與IFN-α+Etoposide 比較,差異有統(tǒng)計(jì)學(xué)意義(P < 0.05)。然而同樣處理MG63 細(xì)胞后結(jié)果顯示,MG63 細(xì)胞在對(duì)照組、IFN-α 組、Etoposide 組和IFN-α+Etoposide 組的細(xì)胞凋亡率分別為:(2.2±0.8)%、(2.5±1.1)%、(23.8±4.6)%和(24.9±5.3)%(圖1B),表明IFN-α 不僅單獨(dú)不誘導(dǎo)明顯的MG63 細(xì)胞凋亡, 也無增強(qiáng)Etoposide 誘導(dǎo)的MG63 細(xì)胞凋亡的作用,Etoposide 組與IFN-α+Etoposide 比較,差異無統(tǒng)計(jì)學(xué)意義(P > 0.05)。
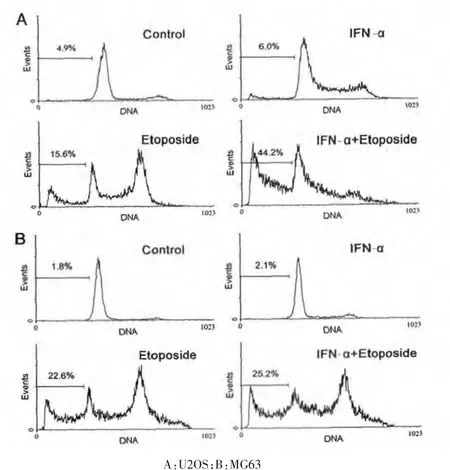
圖1 流式細(xì)胞術(shù)檢測α 干擾素對(duì)依托泊苷誘導(dǎo)的U2OS 和MG63細(xì)胞凋亡的影響
2.2 IFN-α 在U2OS 和MG63 細(xì)胞中對(duì)“DNA 梯形條帶”的影響
U2OS 和MG63 細(xì)胞經(jīng)IFN-α (5000 U/mL)和etoposide (U2OS 細(xì) 胞 為2.5 μg/mL,MG6 細(xì) 胞 為0.625 μg/mL)單獨(dú)或聯(lián)合處理72 h 后進(jìn)行DNA Ladder 電泳。 DNA Ladder 是凋亡的特征性表現(xiàn),結(jié)果顯示, 與其他各組相比,IFN-α/Etoposide 組的U20S 細(xì)胞出現(xiàn)明顯的梯形條帶(圖2A);而在MG63 細(xì)胞,各組均未出現(xiàn)明顯梯形條帶(圖2B)。 表明IFN-α 可明顯增強(qiáng)Etoposide 誘導(dǎo)的骨肉瘤細(xì)胞凋亡, 并且這種效應(yīng)僅出現(xiàn)在p53 功能正常的U2OS 細(xì)胞中,而在因突變導(dǎo)致p53 功能喪失的MG63 細(xì)胞中無此效應(yīng),提示了p53 可能參與此效應(yīng)。
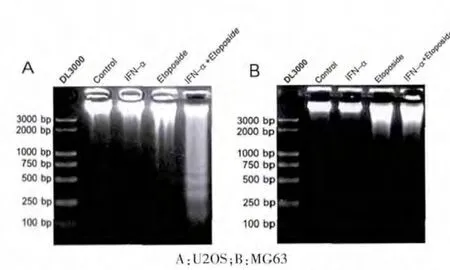
圖2 DNA 梯形條帶分析α 干擾素對(duì)依托泊苷誘導(dǎo)的U2OS 和MG63 細(xì)胞凋亡的影響
2.3 IFN-α 在U2OS 細(xì)胞中對(duì)Etoposide 引起PARP活化的影響
U2OS 細(xì)胞經(jīng)IFN-α (5000 U/mL) 和etoposide(2.5 μg/mL)單獨(dú)或聯(lián)合處理72 h 后進(jìn)行western blot檢測PARP 蛋白活化。 采用Western blot 法檢測凋亡關(guān)鍵酶PARP 的裂解,結(jié)果表明,在U2OS 細(xì)胞,對(duì)照組、IFN-α 組和Etoposide 組均未出現(xiàn)PARP 的活化,在聯(lián)合用藥組可見116 KD 的無活性PARP 前體明顯裂解為85 KD 的活化片段(圖3)。提示聯(lián)合用藥明顯增強(qiáng)PARP 活化,從而觸發(fā)U2OS 細(xì)胞凋亡。
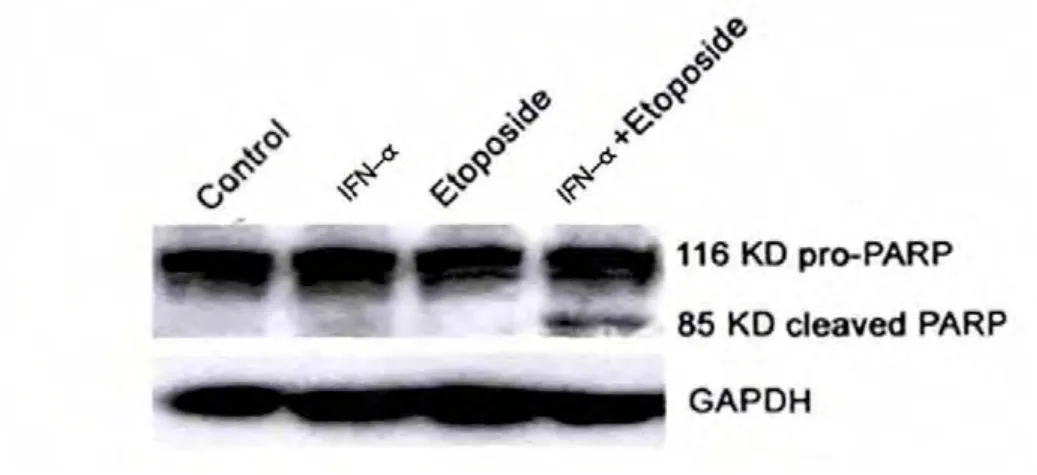
圖3 α 干擾素在U2OS 細(xì)胞中增強(qiáng)依托泊苷誘導(dǎo)的PARP 裂解活化(Western blot 法)
2.4 IFN-α 對(duì)Etoposide 活化p53 凋亡通路的影響
為進(jìn)一步驗(yàn)證IFN-α 對(duì)Etoposide 的增敏作用與野生型p53 的功能有關(guān),筆者進(jìn)一步檢測了p53 凋亡通路相關(guān)基因p53、Bax、Bcl-2、Mdm2 等的表達(dá)。如圖4所示, 在U2OS 細(xì)胞中,IFN-α 單獨(dú)對(duì)p53 及其靶基因MDM2 的表達(dá)無明顯影響, 而Etoposide 可明顯增強(qiáng)二者的表達(dá), 聯(lián)合用藥則進(jìn)一步增高二者的表達(dá)。Bax 和Bcl-2 均是p53 的下游調(diào)控基因, 促凋亡的Bax 的表達(dá)被Etoposide 增高, 并被IFN-α+Etoposide進(jìn)一步增強(qiáng);與對(duì)照組相比,抗凋亡的Bcl-2 的表達(dá)雖然在Etoposide 組無變化,卻被聯(lián)合用藥明顯減弱。因此p53、Bax、MDM2 和Bcl-2 的表達(dá)變化與凋亡保持同步, 提示了在IFN-α 通過激活p53 凋亡通路增強(qiáng)Etoposide 誘導(dǎo)U2OS 細(xì)胞凋亡。 然而在MG63 細(xì)胞中,上述基因的蛋白表達(dá)均無明顯變化,與MG63細(xì)胞中的陰性結(jié)果相符合。
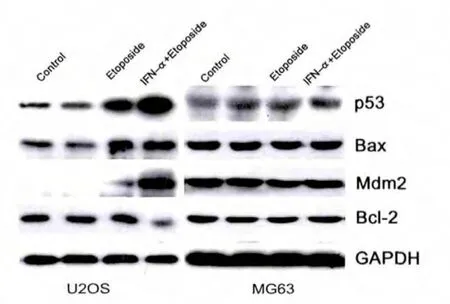
圖4 IFN-α 在U2OS、MG63 細(xì)胞中對(duì)Etoposide 誘導(dǎo)p53,Bax,Mdm2 和Bcl-2 表達(dá)的影響(Western blot 法)
3 討論
干擾素是一類多功能細(xì)胞因子, 具有抗病毒、抗腫瘤和免疫調(diào)節(jié)等多種生物學(xué)作用[1],包括Ⅰ型和Ⅱ型, 其中IFN-α 屬于Ⅰ型干擾素, 可與細(xì)胞表面的IFN-α/β 受體結(jié)合, 從而引起JAK1 和TYK 的活化,活化的JAK1 和TYK 可分別使STAT1 發(fā)生和酪氨酸位點(diǎn)(Tyr701)和絲氨酸位點(diǎn)(Ser727)的磷酸化,磷酸化的STAT1 蛋白形成同源或異源二聚體, 進(jìn)入細(xì)胞核與其靶基因啟動(dòng)子上的干擾素刺激反應(yīng)元件或者γ 活化序列結(jié)合,進(jìn)而調(diào)控下游基因表達(dá)[4]。 IFN-α 也是臨床治療腫瘤的第一種細(xì)胞因子, 已用于膀胱癌、腎癌、肝癌、白血病的臨床治療[5]。
以依托泊苷、阿霉素、順鉑等DNA 損傷性藥物為代表的傳統(tǒng)化療藥物已廣泛用于人類骨肉瘤的新輔助化療,并獲得一定療效,但因其無腫瘤靶向特異性,常常在殺滅腫瘤細(xì)胞的同時(shí)也破壞正常組織細(xì)胞,從而帶來嚴(yán)重的毒副作用,最終導(dǎo)致化療的失敗并影響骨肉瘤的最終療效[6-7]。 有研究表明IFN-α 本身即可抑制多藥耐藥的骨肉瘤細(xì)胞生長[3],那么IFN-α 聯(lián)合化療藥物對(duì)骨肉瘤細(xì)胞的生長是否存在協(xié)同作用,筆者對(duì)此進(jìn)行了一系列研究。
CAD(caspase activated DNAase)是細(xì)胞中一種能切割染色質(zhì)的核酸酶, 可被caspase-3 激活,CAD 可識(shí)別DNA 序列中的特定位點(diǎn),將細(xì)胞基因組DNA在核小體單位之間的連接處剪斷,從而形成180~200 bp或其整數(shù)倍大小的寡核苷酸片段, 經(jīng)瓊脂糖凝膠電泳會(huì)出現(xiàn)梯形電泳條帶,是凋亡的特征性表現(xiàn),因而成為鑒定凋亡的經(jīng)典指標(biāo)[8]。 筆者從流式細(xì)胞術(shù)和形態(tài)學(xué)兩方面證明IFN-α 對(duì)U2OS 細(xì)胞無明顯影響,卻明顯增強(qiáng)Etoposide 誘導(dǎo)凋亡, 提示二者聯(lián)用比化療藥物單用具有更好的抗癌作用。 并且,在U2OS 細(xì)胞中,聯(lián)合用藥可明顯增強(qiáng)PARP[poly(ADP-ribose)polymerase]的活化。實(shí)際上,PARP 是反映凋亡的經(jīng)典指標(biāo),受到促凋亡信號(hào)的刺激后,細(xì)胞內(nèi)蛋白酶Caspases 家族成員caspase 8、9 被激活, 繼而激活下游效應(yīng)性caspase 3,最終裂解其底物PARP 發(fā)生活化,最終導(dǎo)致細(xì)胞凋亡[9]。 上述變化在p53 基因突變而喪失功能的MG63 細(xì)胞中卻沒有發(fā)生,這提示此效應(yīng)可能與p53 有關(guān)。 p53 是一個(gè)經(jīng)典的抑癌基因,在超過50%的惡性腫瘤包括骨肉瘤中都存在缺失或突變, 與腫瘤的發(fā)生發(fā)展關(guān)系密切[10], 作為凋亡調(diào)控的主要靶點(diǎn), 激活的p53 可通過調(diào)節(jié)其下游基因如MDM2,Bax、Bcl-2 等的轉(zhuǎn)錄和表達(dá),誘導(dǎo)細(xì)胞走向凋亡[10]。 目前,p53 已被認(rèn)為是眾多化療藥物(包括Etoposide)抗癌作用的主要介導(dǎo)者[7]。 不僅如此,最近研究表明,p53 也參與了IFN-α 引發(fā)的信號(hào)通路[11]。 筆者發(fā)現(xiàn),Etoposide 激活了p53 及其下游基因如MDM2、Bax 等的表達(dá)。 IFN-α 雖然單獨(dú)應(yīng)用對(duì)p53 通路無明顯影響, 卻明顯增強(qiáng)Etoposide 引起的p53、Bax 和MDM2的表達(dá),同時(shí)減弱了Bcl-2 的表達(dá),充分說明IFN-α通過激活p53 依賴性信號(hào)通路增強(qiáng)Etoposide 誘導(dǎo)的U2OS 細(xì)胞凋亡。
MDM2 基因是調(diào)節(jié)p53 的一個(gè)重要因子, 它在細(xì)胞核中直接與p53 結(jié)合,抑制其轉(zhuǎn)錄活性,并通過胞核-胞漿穿梭將p53 轉(zhuǎn)運(yùn)至胞漿并使其降解,從而降低p53 水平;另一方面,p53 的上調(diào)可激活MDM2轉(zhuǎn)錄表達(dá),來反饋性抑制p53 蛋白的繼續(xù)增高。如此二者形成一個(gè)負(fù)反饋調(diào)節(jié)環(huán), 使細(xì)胞內(nèi)MDM2/p53比率保持恒定[12]。 本研究發(fā)現(xiàn)聯(lián)合用藥引起的p53上調(diào)伴隨著MDM2 水平的升高,表明p53 的上調(diào)反饋性地激活了MDM2 的表達(dá),使其水平升高。Bax 和Bcl-2 基因同屬Bcl-2 家族, 均為p53 的靶基因,表達(dá)于線粒體,是調(diào)控細(xì)胞凋亡的重要基因[13]。二者功能相互拮抗,Bax 具有促進(jìn)凋亡的作用,Bcl-2 則具有抗凋亡的作用, 二者的比例決定了細(xì)胞是否走向凋亡[14-16]。 而聯(lián)合用藥通過激活p53 上調(diào)Bax 和下調(diào)Bcl-2,使Bax/Bcl-2 比例增高,從而促使細(xì)胞發(fā)生凋亡。
因此,IFN-α 可通過誘導(dǎo)p53 依賴性凋亡來增強(qiáng)骨肉瘤U2OS 細(xì)胞對(duì)依托泊苷的敏感性,提示IFN-α與骨肉瘤傳統(tǒng)化療藥物聯(lián)合應(yīng)用可望成為提高骨肉瘤化療療效的新方向。
[1] De Saint SN,F(xiàn)letcher CD.Soft-tissue sarcomas:a update[J].Eur J Surg Oncol,1999,25(2):215-220.
[2] Prejean C,Colamonici OR.Role of the cytoplasmic domains of the type I interferon receptor subunits in signaling [J].Semin Cancer Biol,2000,10(2):83-92.
[3] Manara MC,Serra M,Benini S,et al. Effectiveness of Type I interferons in the treatment of multidrug resistant osteosarcoma cells [J]. Int J Oncol,2004,24(2):365-372.
[4] Stark GR,Kerr IM,Williams BR,et al. How cells respond to interferons[J].Annu Rev Biochem,1998,67(3):227-264.
[5] 郭子文,許曉軍.干擾素輔助小劑量HA 方案治療慢性粒細(xì)胞白血病29 例療效觀察[J].中國醫(yī)藥導(dǎo)報(bào),2012,9(3):37-38.
[6] Bruland OS,Pihl A.On the current management of osteosarcoma. A critical evaluation and a proposal for a modified treatment strategy [J]. Eur J Cancer,1997,33(11):1725-1731.
[7] Hande KR. Etoposide:four decades of development of a topoisomerase II inhibitor [J]. Eur J Cancer,1998,34(10):1514-1521.
[8] Gavrieli Y,Sherman Y,Ben S.Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation [J]. J Cell Biol,1992,119(3):493-501.
[9] Smulson ME,Simbulan-Rosenthal CM,Boulares AH,et al.Roles of poly(ADP-ribosyl)ation and PARP in apoptosis,DNA repair, genomic stability and functions of p53 and E2F-1 [J]. Adv Enzyme Regul,2000,40(2):183-215.
[10] 何均輝,鄒秀玲,李忠楚,等.p53 蛋白在胃癌中的表達(dá)分析[J].中國醫(yī)藥導(dǎo)報(bào),2012,9(5):86-88.
[11] Porta C,Hadj SR,Nejmeddine M,et al.Interferons alphaand gamma induce p53-dependent and p53-independent apoptosis, respectively [J]. Oncogene,2005,24(4):605-615.
[12] Moll UM,Petrenko O. The MDM2-p53 interaction[J]. Mol Cancer Res,2003,1(14):1001-1008.
[13] Bond J,Haughton M,Blaydes J,et al. Evidence that transcriptional activation by p53 plays a direct role in the induction of cellular senescence[J].Oncogene,1996,13(10):2097-2104.
[14] Cory S,Adams JM.The Bcl2 family: regulators of the cellular life-or-death switch[J].Nat Rev Cancer,2002,2(9):647-656.
[15] 黃鳳,毛麗松,張佳.Bcl-2、IGF-2 蛋白在子宮肌瘤組織中的表達(dá)及其相關(guān)性研究[J].中國現(xiàn)代醫(yī)生,2013,51(13):13-14.
[16] 王婷,馬云.Bcl-2 在三陰性乳腺癌中的預(yù)后價(jià)值[J].中國現(xiàn)代醫(yī)生,2012,50(29):44-46.
