心脾綜合征2例并文獻(xiàn)復(fù)習(xí)
2014-04-13 08:53:26楊明烽朱偉華胡麗艷樓楊勇呂小輝陳立軍
浙江醫(yī)學(xué) 2014年16期
楊明烽 朱偉華 胡麗艷 樓楊勇 呂小輝 陳立軍
心脾綜合征2例并文獻(xiàn)復(fù)習(xí)
楊明烽 朱偉華 胡麗艷 樓楊勇 呂小輝 陳立軍
心脾綜合征為一種罕見的發(fā)育異常綜合征,也有學(xué)者稱為內(nèi)臟異位綜合征、異構(gòu)綜合征或側(cè)畸癥(臺(tái)灣地區(qū))[1-2]。本病系由胚胎早期左右不對(duì)稱性分化異常導(dǎo)致的胸腹臟器多發(fā)異常,若趨向正常人體的右側(cè)結(jié)構(gòu)的對(duì)稱化重復(fù)則為無脾綜合征,若為趨向左側(cè)結(jié)構(gòu)的對(duì)稱化重復(fù)則為多脾綜合征[3]。心脾綜合征患者常存在復(fù)雜的先天性心臟病等,因此預(yù)后極差。近年筆者遇到臨床轉(zhuǎn)歸懸殊的2例,現(xiàn)將治療體會(huì)報(bào)道如下。
1 臨床資料
例1 患者女,21歲。2010-07-06因“反復(fù)右上腹疼痛10個(gè)月”入我院肝膽外科治療。體檢:發(fā)育正常,全腹軟,未及壓痛,右上腹觸診飽滿感,壓之不適。本次入院前(2010-06-17)在我院行腹部增強(qiáng)CT檢查示胰腺假性囊腫或囊性占位病變,先天性內(nèi)臟反位,多發(fā)副脾。2010-06-21行上中腹部MRI+MRCP檢查示全內(nèi)臟反位。胰尾部周圍(脊柱右側(cè)脾臟側(cè))囊性占位,考慮胰尾部囊腫可能,先天性胃腸道旋轉(zhuǎn)不良所致腸源性囊腫待排。入院后行X線胸片、心電圖等檢查,符合“右位心”。全科討論考慮腹腔內(nèi)囊腫性質(zhì)可能為畸胎瘤或胰腺囊腺瘤(胰腺真性囊腫)可能,故于2010-07-10全身麻醉下行右上腹腫塊切除術(shù)。取右上腹肋緣下切口,術(shù)中見腹腔內(nèi)臟器反位,各腸段均游離位,屈氏韌帶位于脊柱右側(cè)、回盲部在左下腹部。打開大網(wǎng)膜,見胃后有約5.0cm×5.0cm大小腫塊,呈囊實(shí)性,與胃后壁粘連,胰腺縮小,脾發(fā)育不良。分離粘連,顯露腫塊,發(fā)現(xiàn)腫塊有蒂,并向左側(cè)延伸,與患者的“右肝”(左肋弓部的肝臟)相連,考慮腫塊來源于肝臟,于右肝附著處離斷完整切除。剖視腫塊,見囊性,包膜完整,內(nèi)含淡黃色液體。術(shù)后病理檢查示“右肝”炎性假瘤伴出血壞死部分囊性變。術(shù)后恢復(fù)順利,2010-07-18出院。
術(shù)后1年余,筆者在放射科查閱影像資料時(shí),發(fā)現(xiàn)該例情況特殊,遂細(xì)致研讀并與多位醫(yī)師討論,認(rèn)為應(yīng)考慮多脾綜合征可能。因可能合并先天性心臟病或未明確的其他異常,故電話聯(lián)系患者母親,告知以上情況。2011-10-30,患者及母親來院。詢問病史,除前次手術(shù)情況外無特殊病史,術(shù)后腹痛緩解,現(xiàn)無不適。體檢:無體表畸形,心律齊,心音分布位于右側(cè)鏡像位,未聞及心臟雜音,腹部手術(shù)切口愈合良好。心臟超聲檢查示鏡像右位心,未見其他心臟結(jié)構(gòu)缺陷。再行胸腹部增強(qiáng)CT檢查,細(xì)致閱片后筆者發(fā)現(xiàn)存在以下異常:鏡像右位心,主動(dòng)脈弓及分支為鏡像位,肺靜脈4支引流正常,下腔靜脈腎靜脈水平以下位于左側(cè)發(fā)育正常,右腎靜脈于腹主動(dòng)脈前跨過與左腎靜脈匯合于下腔靜脈。根據(jù)對(duì)比劑分析右腎靜脈血流以進(jìn)入副半奇靜脈為主,左腎靜脈血流以進(jìn)入下腔靜脈為主,腎靜脈和肝靜脈之間的下腔靜脈發(fā)育細(xì)小,下腔靜脈血液主要經(jīng)擴(kuò)張的奇靜脈系統(tǒng)匯入上腔靜脈;左右肺似呈兩葉;腹部?jī)?nèi)臟鏡像反位;門靜脈從胰腺前方行向第一肝門;多脾,位于右側(cè);短胰,呈長(zhǎng)方體形,無胰尾;胃位于右側(cè),向右延伸為十二指腸,從胰腺前上緣、門靜脈后方向后下走行至胰腺左側(cè),十二指腸水平段在胰腺以下水平腸系膜下動(dòng)脈根部從腸系膜上動(dòng)脈后方、腹主動(dòng)脈前方行向右側(cè)后過渡為升段(圖1)。外周血檢查未見Howell-Jolly小體。綜合分析,診斷為“多脾綜合征”。告知患者母親無臟器功能異常,除腸道游離需注意扭轉(zhuǎn)、今后就診需主動(dòng)告知醫(yī)師以上解剖異常外,可正常生活。
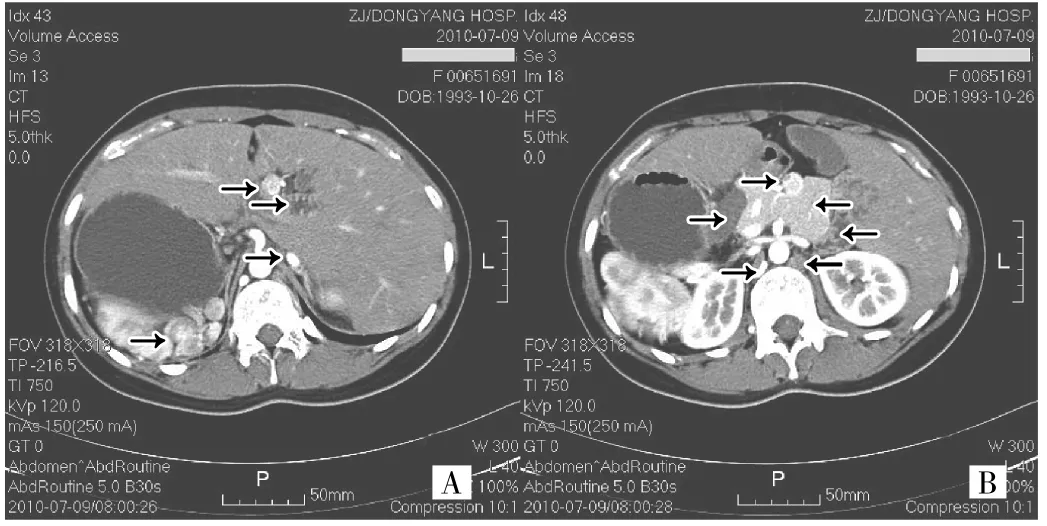
圖1 例1患者腹部增強(qiáng)CT檢查所見(動(dòng)脈期)(A:各箭頭從上至下依次示門靜脈、十二指腸上段、發(fā)育細(xì)小的下腔靜脈、多脾;B:向左箭頭從上至下依次示門靜脈、囊腫、右腎血流主要回流的位于膈肌腳后方的副半奇靜脈,向右箭頭依次示短胰、十二指腸、回流腎臟血流不多的奇靜脈)
例2 患者男,17歲。2012-05-03因“發(fā)現(xiàn)心臟雜音、紫紺14年,胸悶氣促3年”入院。患者出生2個(gè)月后發(fā)現(xiàn)全身紫紺,哭鬧后加重,就診于當(dāng)?shù)匦l(wèi)生院后發(fā)現(xiàn)心臟雜音。后曾在外院就診,心臟超聲檢查提示“復(fù)雜的先天性心臟病”,因手術(shù)風(fēng)險(xiǎn)大,預(yù)后差,故一直未行手術(shù)治療。患者3年前出現(xiàn)活動(dòng)后胸悶氣促,體檢:血壓107/68mmHg,經(jīng)皮血氧飽和度63%,口唇紫紺明顯,手指及腳趾杵狀指/趾明顯,兩肺呼吸音清,未聞及啰音,心前區(qū)隆起明顯,心界無擴(kuò)大,胸骨左上緣可聞及收縮期Ⅲ/6級(jí)噴射樣雜音。入院后心臟超聲檢查示先天性心臟病復(fù)雜畸形:?jiǎn)涡氖遥˙型),肺動(dòng)脈閉鎖,完全性肺靜脈畸形引流(心上型),房間隔缺損(原發(fā)孔+繼發(fā)孔),房室瓣輕度反流,動(dòng)脈導(dǎo)管未閉,體-肺側(cè)支循環(huán)形成。腹部超聲及上腹部CT檢查未見脾臟影像。胸部增強(qiáng)CT檢查示先天性心臟病復(fù)雜畸形,單心室,房間隔缺損,肺動(dòng)脈閉鎖,完全性肺靜脈異位引流(心上型),主動(dòng)脈與房室瓣之間可見肌肉分隔,右位主動(dòng)脈弓,弓上三分支依次為左頭臂干、右頸總動(dòng)脈、右鎖骨下動(dòng)脈,體肺側(cè)支血管形成;細(xì)致分析各臨床資料并討論:經(jīng)超聲和上腹部CT檢查,全腹未及脾臟影像,并結(jié)合血液中發(fā)現(xiàn)Howell-Jolly小體,可確立脾臟先天性缺如;復(fù)雜先天性心臟病情況結(jié)合心臟超聲和CT可明確;外周血檢查發(fā)現(xiàn)Howell-Jolly小體。綜合上述未見脾臟影像及合并復(fù)雜先天性心臟病情況,考慮為無脾綜合征。肺呈兩側(cè)三葉,清晰可見氣管在兩個(gè)水平發(fā)出左上、右上、和左下、右下支氣管;肝臟異常,可分為左右兩半肝,左右各形成3支肝靜脈,并分別匯合成肝上段下腔靜脈,左側(cè)匯入左心房,右側(cè)匯入右心房(圖2)。因患者病情非常復(fù)雜,建議赴上級(jí)醫(yī)院診治。因經(jīng)濟(jì)原因,患者未行進(jìn)一步診治,半年后于家中大咯血死亡。
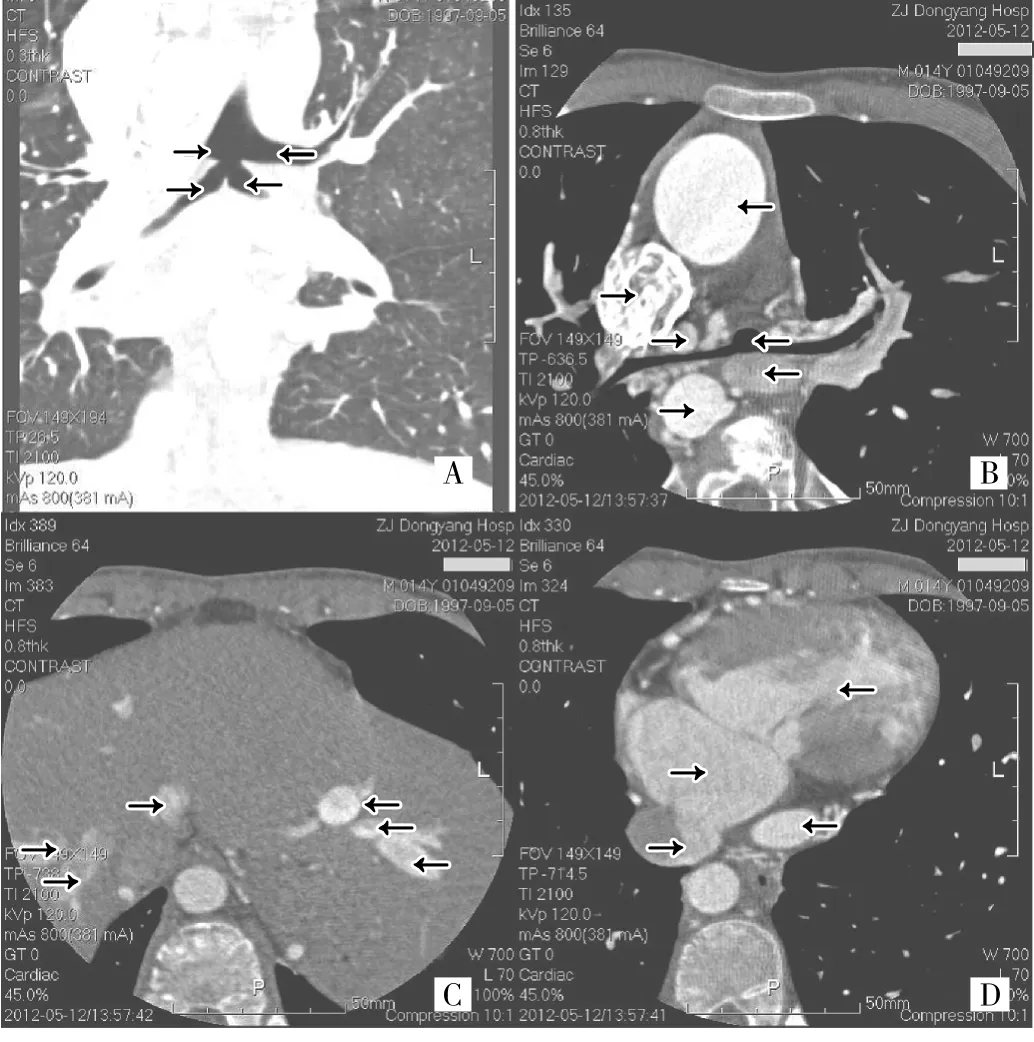
圖2 例2患者的胸部增強(qiáng)CT檢查所見(A:冠狀面重建,向右箭頭從上到下依次示左上、左下支氣管,向左箭頭依次示右上、右下支氣管;B:向左箭頭從上到下依次示上腔靜脈、動(dòng)脈導(dǎo)管、降主動(dòng)脈,向右箭頭依次示升主動(dòng)脈、主氣管發(fā)出左上和右上支氣管處、肺靜脈匯合成的垂直靜脈后匯入左頭臂靜脈;C:向左箭頭示右半肝之三支肝靜脈,向右箭頭依次示左半肝之三支肝靜脈,兩側(cè)肝靜脈造影劑濃度不同;D:向左箭頭從上到下依次示右心房、右肝上段下腔靜脈匯入右心房,向右箭頭依次示單心室、左肝上段下腔靜脈匯入左心房)
2 討論
心脾綜合征作為一種少見疾病,國(guó)內(nèi)除診治先天性心臟病的專科中心有大宗病例報(bào)道以外[4-5],其余以個(gè)案為主,一般醫(yī)務(wù)人員對(duì)該病的認(rèn)識(shí)缺乏,這也是導(dǎo)致本報(bào)道中的2例,影像學(xué)上如此明顯的異常,卻沒有第一時(shí)間發(fā)現(xiàn)或正確描述的原因。
正常人體存在左右不對(duì)稱性,由發(fā)育中的側(cè)化過程導(dǎo)致,若因基因突變,如ZIC3、CRYPTIC[6],或孕婦合并糖尿病妊娠[7]等其他原因?qū)е聜?cè)化異常,則發(fā)育為正常正位人體結(jié)構(gòu)的左右顛倒之鏡相,即為反位,或者介于正位和反位之間的多種變異,即為不定位[3],常導(dǎo)致嚴(yán)重胸腹非成對(duì)臟器的異常,特別是心臟、脾臟、肝臟、腸道、肺等。因心、脾的發(fā)育異常顯著且更有特異性,故常稱不定位患者的復(fù)雜發(fā)育異常為心脾綜合征:若趨向正常人體的右側(cè)結(jié)構(gòu)的對(duì)稱化重復(fù),則為無脾綜合征,表現(xiàn)為無脾、兩側(cè)心耳結(jié)構(gòu)呈寬而鈍三角形的右心耳解剖形態(tài)、雙側(cè)三葉肺等;若為趨向左側(cè)結(jié)構(gòu)的對(duì)稱化重復(fù)則為多脾綜合征,表現(xiàn)為多脾、兩側(cè)心耳結(jié)構(gòu)呈窄而銳指狀的左心耳解剖形態(tài)、雙側(cè)兩葉肺等[8]。心耳結(jié)構(gòu)情況需術(shù)中或解剖所見明確,超聲等影像學(xué)檢查難以明確[3],故臨床診斷主要通過綜合分析各臟器異常作出。
心脾綜合征常合并嚴(yán)重的先天性心臟病,如完全性房室間隔缺損等各種類型的單心室,相對(duì)多脾綜合征,無脾綜合征患者合并的心臟畸形類型常更嚴(yán)重。另無脾綜合征常合并完全性肺靜脈異位引流,多脾綜合征常合并肝段下腔靜脈缺如伴奇靜脈異位引流。心臟異常是此類患者的主要致死原因,另脾臟功能異常、腸旋轉(zhuǎn)不良、膽道閉鎖[8]、呼吸道纖毛功能障礙[9]也是臨床預(yù)后不良因素。
心臟缺陷矯治是治療研究的重點(diǎn),因?yàn)椴∽儑?yán)重,常不能行雙心室矯治,故需行各種Fontan類手術(shù)[1,5,10]。
因治療困難,故產(chǎn)前診斷并終止妊娠則顯得尤為重要,一般通過胸腹臟器是否位置異常、大血管位置、順序節(jié)段法判斷心臟畸形三方面得出診斷[11]。
對(duì)于本文報(bào)道的2例。例1雖為多脾綜合征,但無嚴(yán)重臟器功能異常,存在諸多解剖變異,但無嚴(yán)重臟器功能異常,可正常生活,故對(duì)于產(chǎn)前診斷為多脾綜合征者建議中止妊娠時(shí)應(yīng)慎重考慮。該患者的腹內(nèi)囊腫情況是否與之有關(guān),則有待商榷;患者的下腔靜脈發(fā)育情況,并不符合典型的肝段下腔靜脈缺如伴奇靜脈異位引流,經(jīng)多人判讀,認(rèn)為是文中所述情況,因無臨床必要,故未進(jìn)一步查血管造影。本文例2為無脾綜合征,肺支氣管分支情況特殊,既往國(guó)內(nèi)鮮有報(bào)道;肝靜脈分左右兩組情況既往報(bào)道見于多脾綜合征[12],但分別匯入左右心房則鮮有報(bào)道。
[1]黃繼紅,蘇肇伉,王亮君,等.單心室手術(shù)治療內(nèi)臟異位綜合征早期死亡危險(xiǎn)因素分析[J].上海交通大學(xué)學(xué)報(bào)(醫(yī)學(xué)版),2011,31(9):1269-1271.
[2]林美芳,謝紅寧,李嵐,等.胎兒左、右側(cè)異構(gòu)綜合征產(chǎn)前超聲特征的對(duì)比研究[J].中華超聲影像學(xué)雜志,2011,20(5):432-435.
[3]沈蓉,張玉奇,蔡及明,等.心脾綜合征的多普勒超聲心動(dòng)圖診斷[J].醫(yī)學(xué)影像學(xué)雜志,2007,17(8):836-838.
[4]方敏華,王輝山,朱洪玉,等.Fontan類手術(shù)治療心房異構(gòu)、內(nèi)臟異位合并心臟畸形的效果[J].中華胸心血管外科雜志,2012,28(9):519-521.[5]Belmont J,Mohapatra B,Towbin J A,et al.Molecular genetics of heterotaxy syndromes[J].Curr Opin Cardiol,2004,19(3):216-220.[6]Mart nez-Fras M L.Heterotaxia as an outcome of maternal diabetes:an epidemiological study[J].Am J Med Genet,2001,99(2):142-146.
[7]Serraf A,Bensari N,Houyel L,et al.Surgical management of congenital heart defects associated with heterotaxy syndrome[J].Eur J Cardiothorac Surg,2010,38(6):721-727.
[8]Shiraishi I,Ichikawa H.Human heterotaxy syndrome-from molecular genetics to clinical features,management,and prognosis[J].Circ J,2012,76(9):2066-2075.
[9]Nakhleh N,Francis R,Giese R A,et al.High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy[J].Circulation,2012,125(18):2232-2242.
[10]Jacobs J P,Anderson R H,Weinberg P M,et al.The nomenclature,definition and classification of cardiac structures in the setting of heterotaxy[J].Cardiol Young,2007,17(Suppl 2):1-28.
[11] 石偉元,張彤,張昊晴,等."三步節(jié)段法"產(chǎn)前超聲診斷胎兒內(nèi)臟異位綜合征[J].中國(guó)醫(yī)學(xué)影像技術(shù),2012,28(5):978-981.
[12]劉焦枝.MRI診斷多脾綜合征一例[J].肝膽胰外科雜志,2011,23(6):449,453.
(本文編輯:楊麗)
《浙江醫(yī)學(xué)》對(duì)作者署名的一般要求
同時(shí)具備以下3項(xiàng)條件者方可署名為作者:(1)參與選題和設(shè)計(jì)或資料的分析與解釋者;(2)起草或修改論文中關(guān)鍵性理論或其他主要內(nèi)容者;(3)能對(duì)編輯部的修改意見進(jìn)行核修,在學(xué)術(shù)界進(jìn)行答辯,并最終同意該文發(fā)表者。僅參與研究項(xiàng)目資金的獲得或收集資料者不能列為作者,僅對(duì)科研小組進(jìn)行一般管理者也不宜列為作者。對(duì)文章中的各主要結(jié)論,均必須至少有1位作者負(fù)責(zé)。作者中如有外籍作者,應(yīng)征得其同意,并在投稿時(shí)向編輯部提供相應(yīng)證明材料。集體署名的文稿,在題名下列出署名單位,于文末列出整理者姓名,并須明確該文的主要負(fù)責(zé)人,在論文首頁腳注通信作者姓名、單位、郵政編碼及E-mail地址。通信作者一般只列1位,由投稿者確定。如需注明協(xié)作組成員,則于文末參考文獻(xiàn)前列出協(xié)作組成員的單位及姓名。作者的具體排序應(yīng)在投稿前即確定,在編排過程中不應(yīng)再改動(dòng),確需改動(dòng)時(shí)必須出示單位證明。
本刊編輯部
2013-04-15)
322100 東陽市人民醫(yī)院心臟外科(楊明烽、樓楊勇、呂小輝、陳立軍),放射科(朱偉華),超聲科(胡麗艷)
陳立軍,E-mail:chenlij6606@yahoo.com.cn
