3種頭孢呋辛酯干混懸劑體外溶出行為比較*
2014-05-15 01:42:52林的仕余楚欽林華慶陳潔鄧艷斌林蘇娜吳麗花
醫藥導報 2014年4期
關鍵詞:實驗
林的仕,余楚欽,林華慶,陳潔,鄧艷斌,林蘇娜,吳麗花
(廣東藥學院廣東省藥物新劑型重點實驗室,廣州 510006)
3種頭孢呋辛酯干混懸劑體外溶出行為比較*
林的仕,余楚欽,林華慶,陳潔,鄧艷斌,林蘇娜,吳麗花
(廣東藥學院廣東省藥物新劑型重點實驗室,廣州 510006)
目的 考察3種頭孢呋辛酯干混懸劑(原研、國產、自制)在4種不同溶出介質中的溶出行為,并對其進行相似性評價。方法分別以水、pH 1.2氯化鉀/鹽酸溶液、pH 4.5醋酸鹽緩沖溶液、pH 7.0磷酸鹽緩沖溶液為溶出介質,采用槳法,50 r·min-1進行溶出度實驗。采用紫外分光光度法分別測定不同時間點的溶出量,繪制溶出曲線。以原研頭孢呋辛酯干混懸劑(GlaxoSmithKline)為參比制劑,采用美國食品和藥品管理局(FDA)推薦的相似因子比較法對溶出曲線進行相似性比較。結果自制頭孢呋辛酯干混懸劑在介質為水、pH 1.2氯化鉀-鹽酸溶液、pH 4.5醋酸鹽緩沖溶液、pH 7.0磷酸緩沖溶液中的相似因子分別為74,92,77,75,國產頭孢呋辛酯干混懸劑在以上4種介質中的相似因子分別為40,32,49,50。結論自制頭孢呋辛酯干混懸劑在4種溶出介質中溶出曲線與參比制劑相似,而國產市售頭孢呋辛酯干混懸劑僅在pH 4.5醋酸鹽緩沖溶液和pH 7.0磷酸鹽緩沖溶液中與參比制劑的溶出曲線相似。
頭孢呋辛酯干混懸劑;體外溶出行為;相似性
頭孢呋辛酯(cefuroxime axetil)為第二代頭孢菌素頭孢呋辛的前體藥物。頭孢呋辛酯本身并不具有抗菌活性,口服吸收后迅速被體液和血液中的非特異性酶水解生成頭孢呋辛而發揮抗菌作用[1]。臨床主要用于呼吸道感染、尿道感染、腎盂腎炎、腦膜炎、敗血癥、淋球菌感染等疾病[2]。為滿足臨床上嬰兒、兒童、老年人等不能吞服患者的用藥需要,藥劑科研人員已運用現代制劑技術成功將其制成掩味的干混懸劑,并成功上市。自制頭孢呋辛酯的處方工藝較市售產品有改進,頭孢呋辛酯干混懸劑的標準收載于國家藥品標準WS1-(X-391)-2004Z中[3],但該標準并未涉及溶出度檢查。本品屬低溶解性-高滲透性藥物,溶出是其體內吸收的限速步驟,其與生物利用度密切相關。為評價國產市售頭孢呋辛酯干混懸劑(某醫藥公司)及自制頭孢呋辛酯干混懸劑的內在品質,按照仿制藥一致性評價要求,參考有關文獻及資料[4-6],分別繪制原研(GlaxoSmithKline)、國產、自制3種頭孢呋辛酯干混懸劑在4種不同溶出介質中的溶出曲線,并評價其相似性。
1 儀器與試藥
1.1 儀器 ZRS-8G智能溶出儀(天津市天大天發科技有限公司),ZKT-18F真空脫氣儀(天津市天大天發科技有限公司),UV-1700紫外分光光度計(日本島津公司),KQ-300DA型醫用超聲波清洗機(昆山超聲儀器有限公司),CP225D型、BS224S型電子分析天平[賽多利斯科學儀器(北京)有限公司]。
1.2 試藥 頭孢呋辛酯對照品(中國食品藥品檢定研究院,純度80.8%,批號:130492-200402),頭孢呋辛酯原料藥(廣東立國制藥有限公司,含量:84.1%,批號:FZ41204071),國產頭孢呋辛酯干混懸劑[某醫藥有限公司,規格:0.125 g(C16H16N4O8S),批號: 12060142],原研頭孢呋辛酯干混懸劑[商品名: CEFTIN?,英國GlaxoSmithKline公司,規格:0.250 g (C16H16N4O8S),批號:C600383],自制頭孢呋辛酯干混懸劑[規格0.125 g(C16H16N4O8S),批號: 2013040101]。其他試劑均為分析純。
2 方法與結果
2.1 溶出介質的配制 ①水:為雙蒸水。②pH 1.2氯化鉀/鹽酸溶液:取0.2 mol·L-1的氯化鉀溶液250 mL和0.2 mol·L-1鹽酸溶液425 mL,加水稀釋至1 000 mL,即得。③pH 4.5醋酸鹽緩沖溶液:稱取三水合醋酸鈉2.99 g,加水適量溶解,加2 mol·L-1冰醋酸溶液14.0 mL,并定容至1 000 mL,即得。④pH 7.0磷酸鹽緩沖溶液:稱取無水磷酸氫二鈉5.7 g和磷酸二氫鈉3.7 g,加水溶解并稀釋至1 000 mL,即得。
2.2 對照品溶液的配制 精密稱取頭孢呋辛酯對照品15 mg,置10 mL量瓶中,加甲醇使溶解并稀釋至刻度,搖勻。精密量取1 mL置于100 mL量瓶中,分別加對應溶出介質稀釋至刻度,搖勻,即得。
2.3 專屬性實驗
2.3.1 檢測波長的選擇 取“2.2”項下的對照品溶液,按紫外分光光度法,分別以溶出介質為空白對照,在200~400 nm波長范圍內掃描,繪制對照品溶液紫外掃描圖譜。稱取頭孢呋辛酯干混懸劑適量,置于100 mL量瓶中,加溶出介質適量超聲10 min并稀釋至刻度,搖勻,過濾,取續濾液按紫外分光光度法,分別以溶出介質為空白對照,在200~400 nm的波長范圍內掃描,繪制樣品溶液的紫外掃描圖譜。結果表明,4種不同介質中,對照品與樣品均在280 nm波長處有最大吸收峰出現,故選擇280 nm作為主成分的測定波長。
2.3.2 輔料干擾實驗 按處方量比例,稱取混合輔料適量,以溶出介質為溶劑,制成相當于測定濃度的溶液,過濾,取續濾液,按紫外分光光度法,以溶出介質為空白對照,在200~400 nm的波長范圍內掃描。結果表明,混合輔料在280 nm波長處吸收度干擾不超過2%,可忽略不計。
2.4 線性關系考察 精密稱取頭孢呋辛酯對照品22 mg,置于10 mL量瓶中,加甲醇使溶解并稀釋至刻度,搖勻。精密量取1 mL,置4個10 mL量瓶中,分別用“2.1”項下4種溶出介質稀釋至刻度,搖勻,作為儲備液。分別精密量取儲備液0.2,0.4,0.5,0.6,0.8, 1.0 mL置于10 mL量瓶中,加溶出介質稀釋至刻度,搖勻。按紫外分光光度法,以溶出介質為空白,于280 nm處測定吸光度。以吸光度(A)對濃度(C)進行線性回歸。結果表明,頭孢呋辛酯在3.61~18.05 μg·mL-1范圍內,在280 nm處吸光度與濃度呈良好線性關系。見表1。
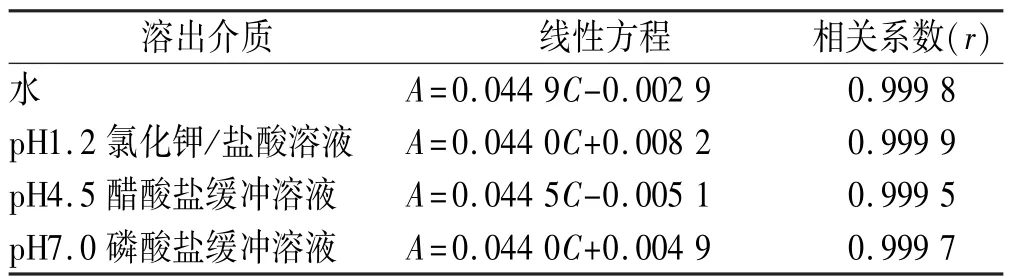
表1 頭孢呋辛酯在4種不同pH介質下的線性方程與相關系數
2.5 濾膜吸附實驗 工作中常用的濾膜分為水系和有機系兩種,因選取的4種介質均不含有機溶劑,根據《中華人民共和國藥典》2010年版二部相關要求并結合樣品實際情況,確定選取水系膜,孔徑為0.8 μm (φ=25 mm)進行濾膜吸附驗證實驗。取“2.2”項下的對照品溶液適量,將其分為兩份,一份不過濾,另一份用濾膜過濾,在280 nm處測定兩者的吸光度。結果表明,濾膜吸附量均<2%,可忽略不計[7]。
2.6 精密度實驗 分別精密量取“2.4”項下的儲備液0.4,1.2,2.0 mL,置于20 mL量瓶中,加溶出介質稀釋至刻度,搖勻。以溶出介質為空白,于280 nm波長處測定吸光度,各連續測定6次。見表2。
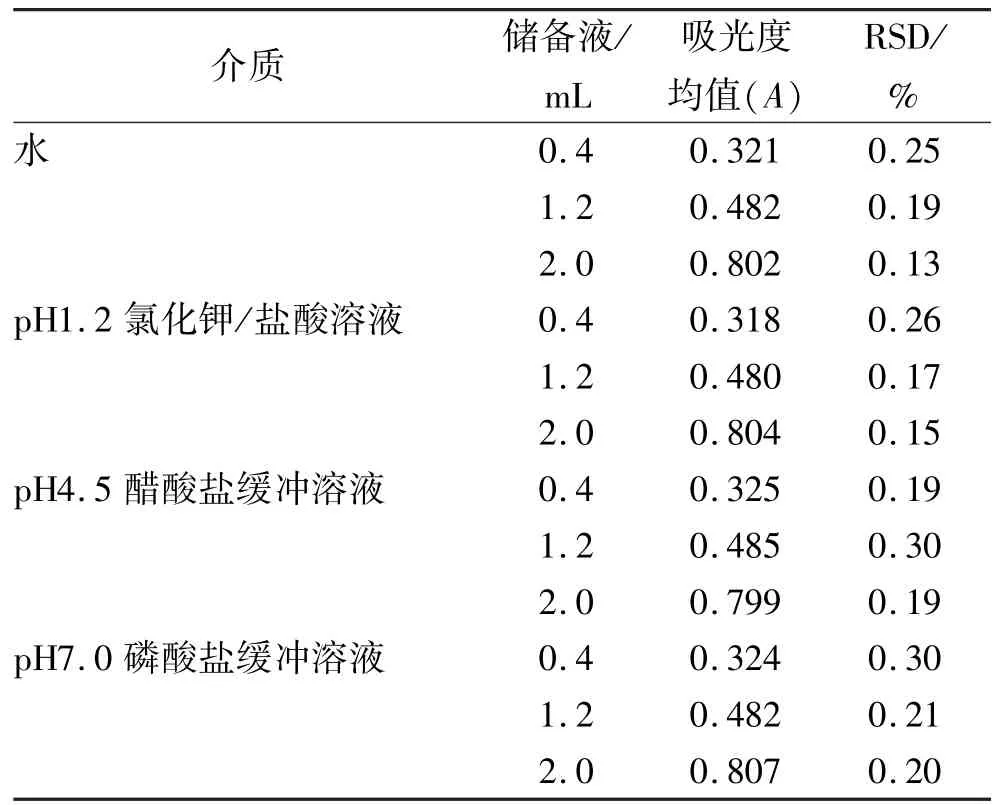
表2 4種介質精密度實驗結果
2.7 重復性實驗 取頭孢呋辛酯干混懸劑(批號: 2013040101)加水適量配制成規格為25 mg·mL-1(以頭孢呋辛計)的混懸液,取以上混懸液5 mL,每次6份,共測定3次。依溶出度測定法(漿法),實驗溫度: 37℃,分別以水、pH 1.2氯化鉀/鹽酸溶液、pH 4.5醋酸鹽緩沖溶液、pH 7.0磷酸鹽緩沖溶液900 mL為溶劑,轉速為50 r·min-1,依法操作,經5,15,90 min時(其中水、pH4.5的介質取樣點為5,30,360 min),取溶液5 mL濾過,棄去初濾液,精密量取續濾液適量,加對應溶出介質稀釋制成每毫升中含11 μg的溶液,作為供試品溶液。取上述溶液及“2.2”項下的對照品溶液,照紫外-可見分光光度法,分別在280 nm波長處測定吸光度,計算不同時間的溶出量。見表3。
2.8 溶液穩定性實驗 取頭孢呋辛酯干混懸劑置研缽充分研磨,分別稱取4份置于100 mL量瓶中,分別加溶出介質適量,超聲15 min使溶解并稀釋至刻度,搖勻,過濾,各取濾液適量,于室溫放置,并于0,0.5, 1,2,3,4,5和6 h測定吸光度。結果表明,測定溶液在室溫下,6 h內的RSD均<2%,溶液基本穩定。
2.9 加樣回收率實驗 精密稱取已知含量(31.25 mg·g-1,以頭孢呋辛計)的頭孢呋辛酯干混懸劑樣品9份,每份0.1 g,置100 mL量瓶中,分別精密加入頭孢呋辛酯對照品適量(以頭孢呋辛計),先加甲醇適量超聲溶解,再加溶出介質稀釋至刻度,搖勻,濾過,精密量取2 mL置于100 mL量瓶中,分別加對應溶出介質稀釋至刻度,搖勻,照紫外分光光度法,在280 nm處測定吸光度。另取“2.2”項下對照品溶液,同法測定,計算回收率。見表4。
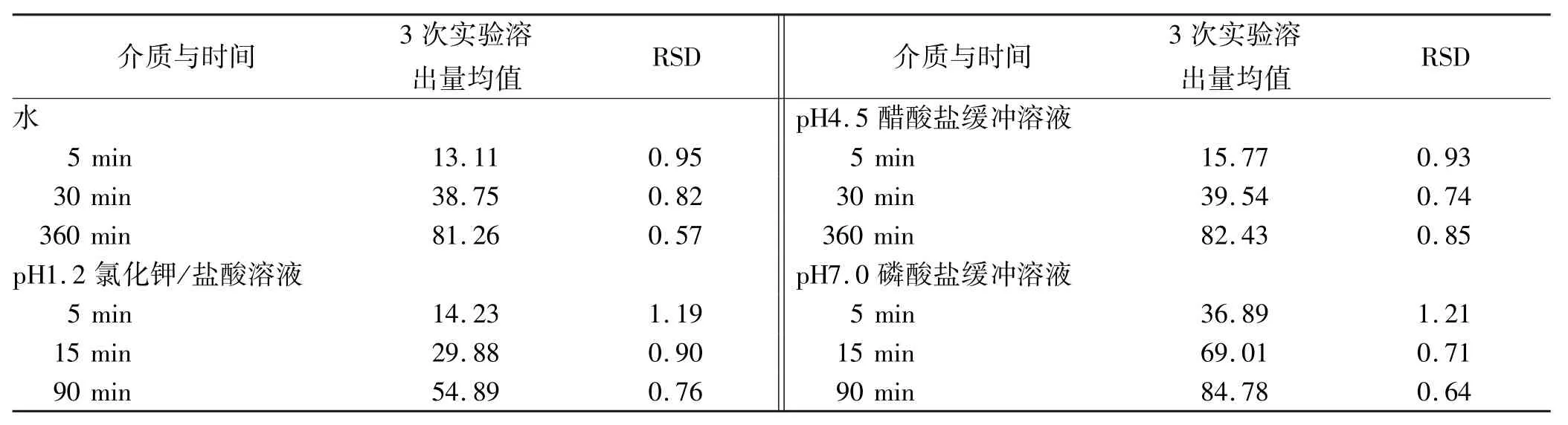
表3 4種介質重復性實驗結果 %
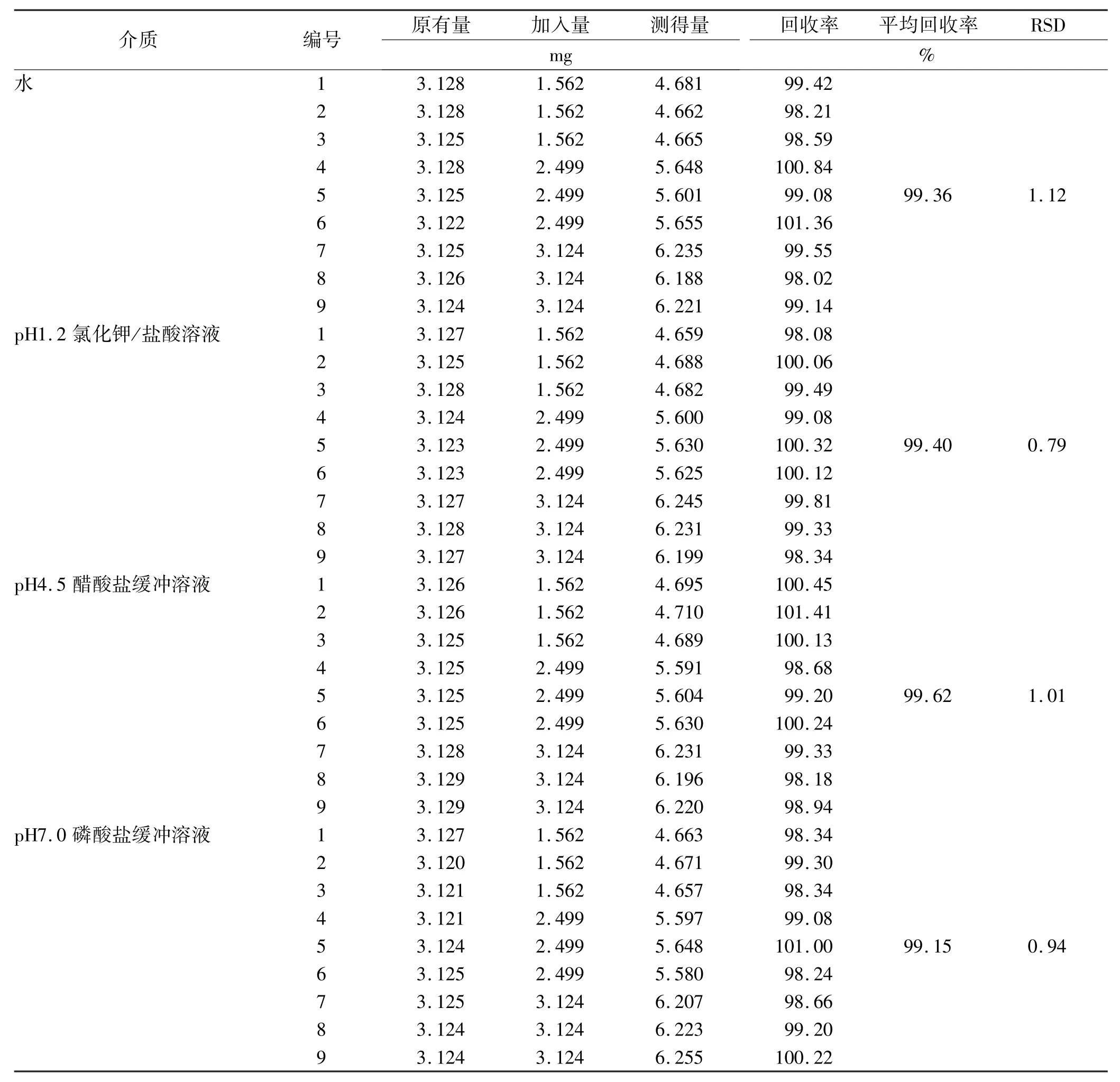
表4 頭孢呋辛酯在4種介質下的加樣回收率實驗結果
2.10 溶出曲線繪制 按如下方法[4]分別測定原研、國產、自制3種頭孢呋辛酯干混懸劑在不同pH的溶出介質中的溶出曲線,各測定3次:取頭孢呋辛酯干混懸劑加水適量配制成規格為25 mg·mL-1(以頭孢呋辛計)的混懸液。取上述混懸液照溶出度測定法(漿法),實驗溫度:37℃,分別以水、pH 1.2氯化鉀/鹽酸溶液、pH 4.5醋酸鹽緩沖溶液、pH 7.0磷酸鹽緩沖溶液900 mL為溶劑,轉速為50 r·min-1,依法操作,經5,10,15,30,45,60,90,120,180,240,300,360 min時 (pH1.2、pH7.0的介質則取樣至120 min時結束實驗),取溶液5 mL濾過(同時補充同溫介質5 mL),棄去初濾液,精密量取續濾液適量,加溶出介質稀釋制成每毫升中含11 μg的溶液,作為供試品溶液。取上述溶液及“2.2”項下的對照品溶液,照紫外-可見分光光度法,分別在280 nm波長處測定吸光度,計算不同時間的累積溶出百分率。以時間對溶出百分率作圖,繪制溶出曲線。見圖1。

A.水;B.pH 1.2氯化鉀/鹽酸溶液;C.pH 4.5醋酸鹽緩沖溶液;D.pH 7.0磷酸鹽緩沖溶液圖1 頭孢呋辛酯干混懸劑在4種介質中的溶出曲線(n=3)
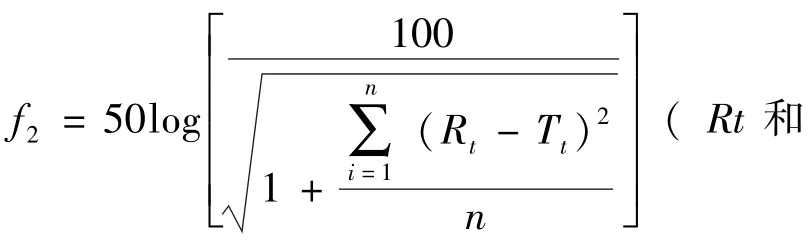
結果可見與參比制劑相比自制頭孢呋辛酯干混懸劑在4種不同pH介質條件下相似因子f2均>46,說明體外溶出行為相似。而國產頭孢呋辛酯干混懸劑在pH4.5醋酸鹽緩沖溶液條件下相似因子f2>46,在 pH7.0的磷酸鹽緩沖溶液條件下相似因子f2>42,說明在這兩種溶出介質中溶出行為相似。在水、pH1.2氯化鉀/鹽酸溶液中溶出行為不相似。見表5。

表5 4種溶出介質條件下溶出曲線相似因子f2
3 討論
頭孢呋辛酯干混懸劑的國家藥品標準WS1-(X-391)-2004Z中并未有溶出度質量標準,根據《普通口服固體制劑溶出度實驗技術指導原則》,采用《美國藥典》中該品種的溶出測定方法進行不同溶出介質條件下溶出曲線的比較[8]。結果顯示溶出較為緩慢,但在規定時間內已有一定落差,不影響溶出曲線的相似性評價,且由于該品種主要是嬰兒、兒童、老年人以及其他不能吞服的患者服用,一般認為槳法50 r·min-1與這類患者消化道的蠕動強度相當,故不再放寬實驗條件。
文獻[9]指出參比制劑溶出率在規定時間內達85%以上,以參比制劑平均溶出率達85%的時間點為Ta,比較1/4Ta、1/2Ta、3/4Ta和Ta 4個時間點的兩者平均溶出率f2因子>42;達50%~85%或85%以上的,以參比制劑在結束時間點平均溶出率的85%的時間點作為Ta,比較1/4Ta、1/2Ta、3/4Ta和Ta 4個時間點,f2因子>46,則可判定為相似。本研究中各計算時間點數據偏離度均符合要求。
國內已有溶出度標準的干混懸劑溶出實驗時,多采用直接投入干混懸劑的方法。筆者也嘗試該投樣方法進行實驗,發現除在pH 7.0磷酸鹽緩沖溶液中頭孢呋辛酯干混懸劑在轉漿攪動下能分散在介質中外,其他3種溶出介質均是漂浮在液面,影響溶出。故最終采取第35版《美國藥典》中該品種的投樣方法。相比之下,采取投入混懸液的方法更能真實模擬服藥過程,有助于建立體內外相關性。
國產上市頭孢呋辛酯干混懸劑批內均一性和批間重復性均良好,與參比制劑的溶出曲線卻存在差異,說明其內在品質與參比制劑仍存在差異。參比制劑與自制制劑兩者在4種溶出介質條件下溶出曲線均相似,初步認定自制制劑處方、工藝合理,具有與與參比制劑相當的內在品質,但它僅在一定程度上預測生物等效性的研究情況及指導生物等效性實驗受試者的選擇,其生物等效性和臨床療效有待生物等效性實驗和臨床實驗驗證。
[1]HARDING S M,WILLIAMS P E O,AYRTON J.Pharmacology of cefuroxime as the 1-acetoxyethylester in volunteers[J].Antimicrob Agents Chemother,1984,25 (1):78-82.
[2] GOOCH W M,SWENSON E,HIGBEE M D,et al.Cefuroxime axetil and penicillin V compared in the treatment of group A beta-hemolytic streptococcal pharyngitis[J].Clin Ther,1987,9(6):670-677.
[3] 國家藥典委員會.國家藥品標準新藥轉正標準第60冊[S].北京:人民衛生出版社,2005:44-45.
[4] U.S.Pharmacopeia 35/National Formulary 30[S].The United States Pharmacopeial Convention,2011:2579.
[5] 潘強,潘智鵬,徐秋陽,等.頭孢呋辛酯干混懸劑溶出度測定方法的建立[J].中國藥事,2012,26(1):66-67.
[6] PMDA.後発醫薬品の生物學的同等性試験ガイドラインについて[EB/OL].http://www.pmda.go.jp,2013-01-10.
[7] 康建磊,徐冰珠.速釋口服固體制劑溶出度研究驗證中需注意的問題[J].解放軍藥學學報,2010,26(4):369-373.
[8] 謝沐風.溶出曲線的測定與比較[R].上海:上海市藥品檢驗所,2008.
[9] 謝沐風.溶出曲線相似性的評價方法[J].中國醫藥工業雜志,2009,40(4):308-310.
DOI 10.3870/yydb.2014.04.028
R978.11;TQ460.6
A
1004-0781(2014)04-0503-05
2013-06-05
2013-09-28
*藥物制劑關鍵技術研發及產業化示范項目(2008A1-E4101)
林的仕(1988-),男,廣東揭陽人,碩士,主要從事緩控釋制劑的研究。電話:(0)13450207406,E-mail:lindishi1988@163.com。
林華慶(1964-),男,廣東湛江人,教授,碩士生導師,學士,主要從事緩控釋制劑和中藥新劑型的研究。電話: 020-39352502,E-mail:huaqing_@vip.tom.com。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
