聚吡咯改性電沉積Sn負極材料的電化學性能
2014-07-14 05:19:38溫兆銀吳梅芬楊建華
無機化學學報 2014年6期
彭 鵬 溫兆銀 劉 宇 吳梅芬 楊建華
(中國科學院上海硅酸鹽研究所能量轉換重點實驗室,上海 200050)
0 引 言
鋰離子電池因具有能量密度高、重量輕、環境友好等優點已廣泛應用于電子器件市場。Sn因能與鋰形成Li22Sn5儲鋰產物,具有比商用石墨電極(372 mAh·g-1)高得多的比容量(987 mAh·g-1)[1,2]而引起了廣泛的關注。該類材料還具有脫嵌鋰電壓低(ca.0.8 V vs Li/Li+)、與電解液的反應活性低等優點,有利于發展成為大功率安全性電源之一。Sony公司推出的Sn-Co-C鋰離子電池進一步推動了錫基合金負極材料的研究,但這類材料在電化學脫嵌鋰過程中存在著較大的體積效應,產生的機械應力會使電極活性物質破碎,喪失與集流體的電接觸,從而造成電極電化學循環性能的衰減[3]。目前解決該問題的方法有很多,包括引入其他活性或者惰性相形成合金體系或者其他導電材料形成復合體系等。Tabuch等[4]通過共電鍍的方式得到Sn-Sb-Co三相合金,具有580 mAh·g-1的可逆比容量,其他合金體系還有Sn-Cu[5]、Sn-Sb[6,7]、Sn-Ag[8]、Sn-Ni[9]、Sn-Ca[10]等。Wang 等[11]將Sn分散在有序介孔石墨碳層中,利用碳層骨架形成的有序導電網絡緩解充放電過程中的體積效應,100次循環后剩余560 mAh·g-1穩定比容量。本工作在銅箔上直接電沉積Sn負極,避開常規操作中粘結劑的使用,簡化工藝程序,同時利用導電聚合物聚吡咯(PPy)對沉積后的電極片進行表面修飾改性,聚吡咯具有良好的彈性和粘附性,可形成穩定牢固的導電通道[12-13],能有效緩解合金負極的體積效應同時增加材料的導電性,保持活性材料與集流體的電接觸,改善Sn負極的電化學性能。
1 實驗部分
1.1 電極材料的制備
1.1.1 Sn 負極的電沉積
以厚度15 μm銅箔為工作電極,厚度150 μm鎳片為對電極,在自制簡易電解槽(如圖1)中恒電流密度(0.35 mA·cm-2)恒溫(50 ℃)沉積 2h,電解液為SnCl2、NaF(各 30 g·L-1,鹽酸調節 pH=4~5),取出電極,經去離子水洗滌、干燥,簡記為Sn。
不同濃度 NiCl2(0、10、25、50、100 g·L-1) 作為添加劑分別溶解于SnCl2-NaF電解液中,重復相同的步驟,沉積10 min,電極經去離子水洗滌、干燥,分別簡記為 Sn(Ni)-0、Sn(Ni)-10、Sn(Ni)-25、Sn(Ni)-50、Sn(Ni)-100。
1.1.2 聚吡咯修飾Sn的電極制備
經去離子水洗滌的電極懸掛浸入預先超聲分散均勻的吡咯懸浮液中,0℃逐滴緩慢加入氧化劑(NH4)2S2O8(n吡咯:n(NH4)2S2O8=1∶1),滴加完畢后靜置 3 h,取出電極,經去離子水洗滌、干燥。
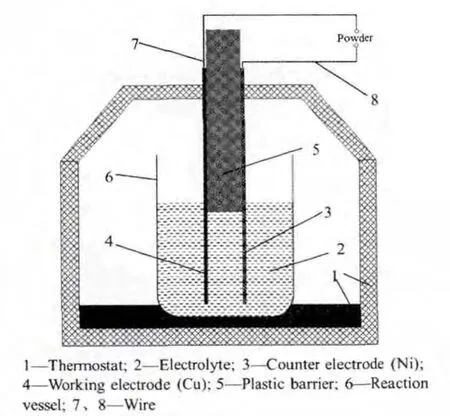
圖1 自制簡易電解槽示意圖Fig.1 Schematic diagram of the electrolysis setup
1.2 電極材料的表征及電化學性能測試
材料的結構由X射線衍射(Riguka,D/max2550VB3+/PC 型,Cu 靶,Kα1射線,λ=0.154 056 nm,掃描電壓 40 kV,電流 100 mA,掃描范圍 10°~80°,掃描速度為 6°·min-1)表征,JSM-6700 型掃描電子顯微鏡分析電極片的表面形貌;元素分布由INCA型能譜儀分析(英國牛津)。將沉積的電極片直接切成圓片(φ 14 mm),以金屬鋰為對電極和參比電極,Celgard2325 為隔膜,1 mol·L-1LiPF6的碳酸乙烯酯 (ethylene carbonate,EC):碳酸二甲酯(dimethyl carbonate,DMC)(物質的量的比為 1∶1)有機溶液(其中含有2wt%碳酸亞乙烯酯)為電解質,組裝成2025扣式電池。LAND CT2001A充放電儀恒流模式進行充放電,電流密度為120 mA·g-1,電壓范圍為0.1~1.5 V。Autolab PGSTAT302型電位-恒電流電化學工作站分析電化學循環伏安曲線和電化學交流阻抗譜,循環伏安的掃描電壓范圍為 0.05~1.5 V,掃描速度為0.5 mV·s-1,交流阻抗譜測試頻率范圍為10-2~105Hz,交流振幅為5 mV。
2 結果與討論
2.1 電解液成分的影響
圖2顯示的是在電解液中添加了不同濃度NiCl2后沉積電極的XRD圖,除去銅箔的特征衍射峰 ,Sn(Ni)-10、Sn(Ni)-25、Sn(Ni)-50、Sn(Ni)-100 都 在30.3°出現了對應于四方結構 Sn(a=b=0.538 1 nm,c=0.318 72 nm,PDF No.04-0673)的(200)特征衍射峰。所有沉積電極的XRD圖中均沒有雜相衍射峰,說明電極表面是純相Sn。而沒有添加NiCl2的沉積電極Sn(Ni)-0的XRD圖中沒有Sn的特征衍射峰出現,說明在短時間內電極表面沒有Sn顆粒析出。添加了NiCl2有利于縮短Sn負極的沉積時間,提高了沉積時率。
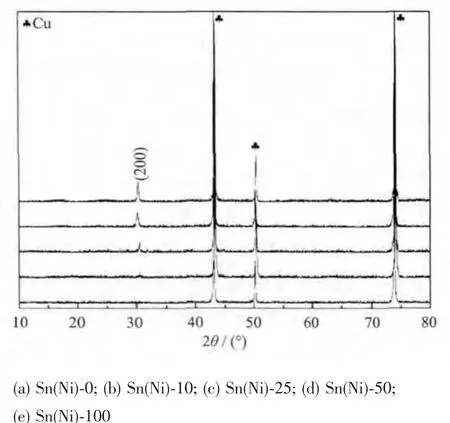
圖2 不同濃度NiCl2沉積10 min的電極XRD圖Fig.2 XRD patterns of anodes deposited for 10 min with different concentrations of NiCl2
在電解液中添加不同濃度NiCl2后沉積10 min的電極形貌如圖3所示。比較發現,電解液中沒有添加NiCl2沉積的電極表面沒有Sn顆粒析出,而添加了NiCl2的4種電解液中,沉積電極的表面均析出了一層細小的納米Sn顆粒同時伴隨著類似Sn空心管顆粒。隨著NiCl2濃度增加,電極表面析出的Sn顆粒越多顆粒粒徑隨之長大,空心管也逐漸變大。這是因為NiCl2濃度越大,析出Sn顆粒速率越快,沉積在銅箔表面的Sn顆粒生長也越快,因而粒徑越大。但是當NiCl2濃度過高,例如Sn(Ni)-100的表面形貌,不僅表面沉積的納米Sn顆粒粒徑分布不均,甚至Sn空心管開始消失,而一端開口的球形顆粒開始增多,這是因為NiCl2濃度過高導致Sn顆粒在電極表面析出速率過快,顆粒來不及自組裝成空心管,更容易團聚成球,這對利用Sn空心管狀結構來緩解體積效應是不利的。

圖3 不同濃度NiCl2沉積10 min的電極表面SEM圖Fig.3 SEM images of anodes deposited for 10 min with different concentrations of NiCl2
圖4中對應的EDS圖也顯示添加了不同濃度NiCl2沉積的電極表面EDS相似,都存在Sn元素的峰,而沒有添加NiCl2沉積的電極Sn(Ni)-0表面沒有Sn元素峰存在。此外EDS圖中除了銅箔對應的Cu元素以及導電膠的C元素外,沒有Ni元素峰出現,說明電極表面顆粒是純相Sn。
值得注意的是EDS譜中都沒有干擾元素F出現,說明NaF不參與電沉積反應。SnCl2溶于水易分解,NaF可與SnCl2形成配合物[SnClF2]增加溶解度,同時能降低Sn2+的析出電位。此外,在鎳片為對電極的沉積體系中,電解液中添加NiCl2有助于抑制鎳對電極發生不必要的副反應進入電解液中,從而影響沉積效果。
2.2 聚吡咯表面修飾
結合沉積時效和沉積電極表面形貌,選擇Sn(Ni)-0、Sn(Ni)-50沉積 2 h,繼續研究 NiCl2對沉積電極電化學性能的影響,對應電極分別簡記為Sn-2h、Sn(Ni)-2h,同時分別對沉積電極的表面進行聚吡咯修飾,簡記為 Sn-2h-PPy、Sn(Ni)-2h-PPy。
2.2.1 沉積電極的表面形貌表征
圖5為沉積電極和聚吡咯修飾后電極表面的SEM圖,圖5(a)中Sn-2h析出的細小納米顆粒均勻分布在銅箔基體表面,圖5(b)中Sn(Ni)-2h表面呈現空心管周圍伴隨著微米級球狀顆粒的形貌,空心管內徑約 2.5 μm,外徑約 4.0 μm,長度在 20 μm 以上。聚吡咯修飾后,Sn-2h-PPy電極表面覆蓋了一層粒徑分布集中且分散性好的球狀聚吡咯納米顆粒(粒徑小于500 nm),Sn(Ni)-2h-PPy電極表面的空心管也覆蓋著球狀聚吡咯納米顆粒。
2.2.2 電化學性能研究
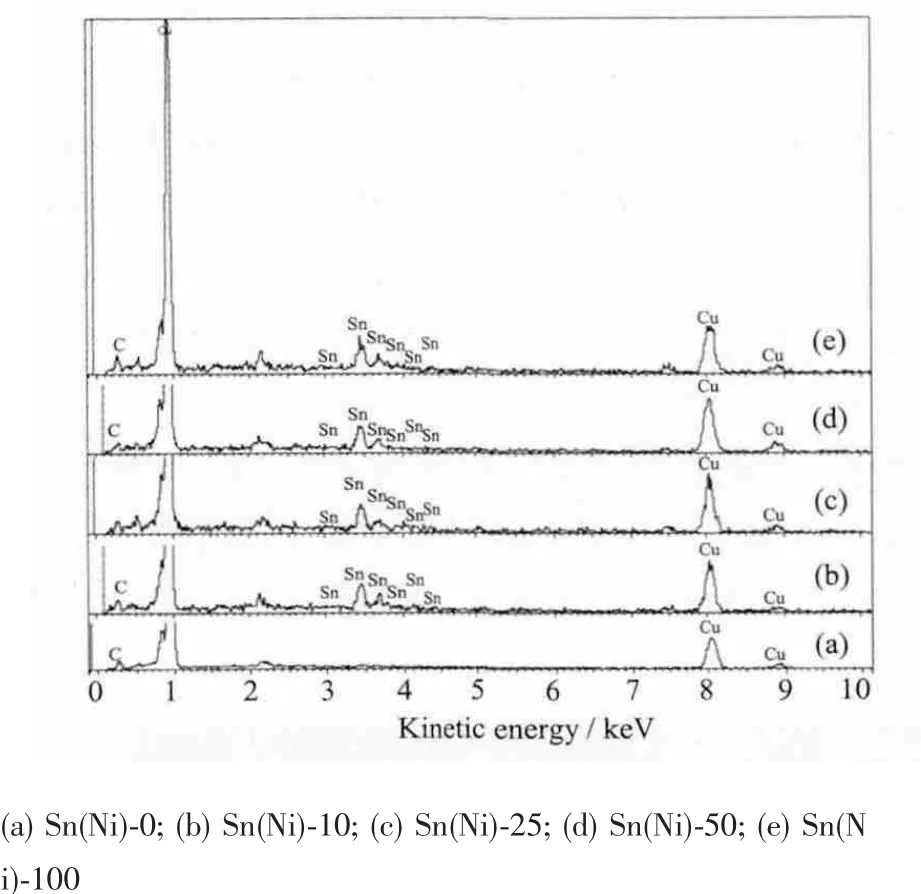
圖4 不同濃度NiCl2沉積10 min的電極表面EDS圖Fig.4 EDS of anodes deposited for 10 min with different concentrations of NiCl2
圖6(a)循環壽命曲線顯示Sn-2h在首次循環后可逆比容量迅速衰減殆盡,且首次放電比容量也較低,這是由于納米級Sn顆粒單獨分散在銅箔基體表面,相互間的作用力小,周圍沒有其他相能夠提供保護效用,緩解循環過程中合金化/去合金化產生的巨大體積效應,從而造成Sn顆粒發生破碎,從基體上脫落,喪失與集流體的電接觸,使得容量迅速衰減。Sn(Ni)-2h電極由于微米級球狀顆粒的大量存在,這些顆粒的體積效應不能被自身的結構所消化,容易破碎失去脫嵌鋰活性,但是另外由于Sn空心管結構的存在,這種結構對體積效應存在一定程度的緩解作用,即使在沒有其他相的輔助保護下,仍然能保有一些電化學活性,故Sn(Ni)-2h電極在60次循環后仍剩余一部分可逆比容量,達184.3 mAh·g-1。
利用PPy表面修飾后Sn-2h-PPy、Sn(Ni)-2h-PPy電極的電化學循環穩定性能都有了大幅度的提升。在60次循環后分別剩余397.5、440.6 mAh·g-1可逆比容量。
從圖6(a)的內插圖可看出雖然Sn-2h-PPy的可逆比容量在循環初期高于Sn(Ni)-2h-PPy,但隨著循環次數的增加可逆比容量呈現小幅度緩慢衰減的變化趨勢,在57次循環后可逆比容量已經低于Sn(Ni)-2h-PPy了且仍沒達到穩定平衡態,而Sn(Ni)-2h-PPy從30次循環以后可逆比容量基本保持穩定,因此Sn(Ni)-2h-PPy電極在保持高可逆比容量的同時循環穩定性也非常優越,說明表面修飾了聚吡咯對電極的電化學循環性的確有一定的改善,能夠提高循環的穩定性。

圖5 沉積電極和修飾電極表面的SEM圖Fig.5 SEM images of deposited and modified anodes

圖6 沉積電極和修飾電極的循環壽命曲線(a)和充放電曲線(b)Fig.6 (a)Capacity vs.cycle number of deposited and modified anodes;(b)Potential vs.Capacity of deposited and modified anodes
圖6(b)中首次充放電曲線顯示Sn-2h-PPy首次放電比容量為2 149 mAh·g-1,首次庫倫效率為35.3%,Sn(Ni)-2h-PPy首次放電比容量提高至3 252 mAh·g-1,首次庫倫效率高達 68.8%,很明顯,Sn(Ni)-2h-PPy首次庫倫效率更高。分別比較兩種材料的第30、60次充放電曲線,Sn(Ni)-2h-PPy電極的對應充放電曲線基本重合,而Sn-2h-PPy出現較大程度的衰減,說明Sn(Ni)-2h-PPy電極的循環穩定性更好。
聚吡咯位于沉積電極表面可作為保護膜減少電極表面活性物質與電解液的副反應,同時其良好的彈性可抑制體積效應引起的活性材料破碎,增加脫嵌鋰過程可逆性,提高電極材料的循環穩定性。
圖7為Sn(Ni)-2h-PPy電極的循環伏安曲線,是典型的Sn負極脫嵌鋰過程。在還原曲線上位于0.65、0.75、0.8 V 的峰表示 Sn 的去合金過程,對應的合金化過程的峰出現在氧化曲線的 0.35、0.65 V,說明Sn(Ni)-2h-PPy電極的脫嵌鋰過程是分步進行的[14-15]。沒有對應于PPy脫嵌鋰過程的峰出現,說明在0.05~1.5 V的電壓范圍PPy不參與脫嵌鋰反應,對電極材料的容量沒有貢獻[16].
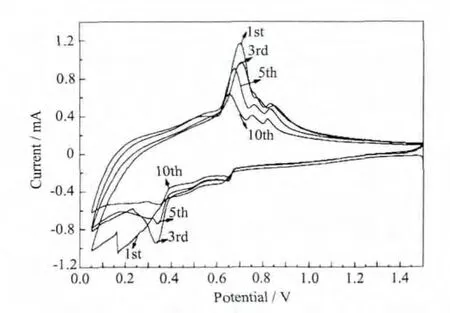
圖7 Sn(Ni)-2h-PPy電極的循環伏安曲線Fig.7 Cyclic voltammogram of Sn(Ni)-2h-PPy electrode
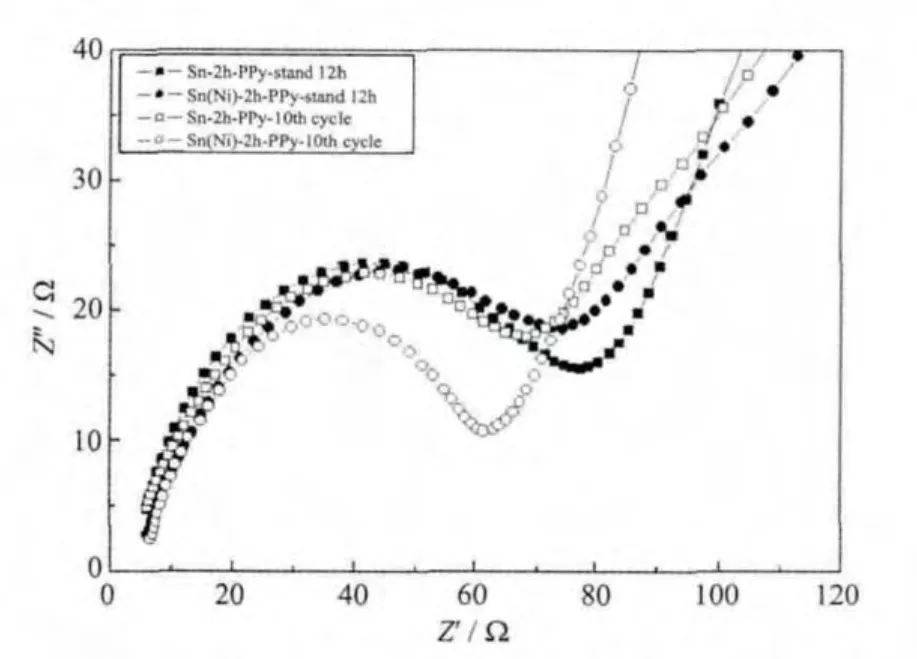
圖8 修飾電極循環前后的阻抗譜Fig.8 EIS spectra of modified anodes before and after 10 cycles
圖8為PPy修飾后電極循環前后的阻抗圖。中高頻區的半圓對應于電極的本征電子電阻和接觸電阻以及金屬鋰對電極的表面鈍化膜電阻[22-24],低頻區斜線的斜率約為45°,代表Warburg阻抗[25]。循環后修飾電極在中高頻區的阻抗都減小,說明電極經歷了活化過程,開始展現優越的電化學性能。而Sn(Ni)-2h-PPy電極在經歷10次循環后中高頻區的阻抗明顯比Sn-2h-PPy小得多,根據阻抗越大,則電導率越低,對應極材料的電化學性能越差,側面證明了Sn(Ni)-2h-PPy電極的循環性能更穩定。
3 結 論
通過在銅箔上電沉積直接得到Sn負極,避開了粘結劑的使用。以NiCl2為沉積電解液的添加劑得到了Sn空心管,提高了材料的可逆比容量。進一步引入聚吡咯進行沉積電極表面修飾改性,有效地提高了電極的電化學循環穩定性,得到首次庫倫效率高達 68.8%,60 次循環仍剩余 440.6 mAh·g-1可逆比容量的負極材料。
[1]Ng S H,Wang J Z,Wexler D,et al.Angew.Chem.Int.Ed.,2006,45(41):6896-6899
[2]Simon G K,Goswami T.Metall.Mater.Trans.,2011,42A:231-238
[3]Kim H,Han B,Choo J,et al.Angew.Chem.Int.Ed.,2008,47(52):10151-10154
[4]Tabuchi T,Hochgatterer N,Ogumi Z,et al.J.Power Sources,2009,188(2):552557
[5]Wolfenstine J,Campos S,Foster D,et al.J.Power Sources,2002,109:230-233
[6]Yang J,Takeda Y,Imanishi N,et al.Solid State Ionics,2000,135:175-180
[7]Zhao H L,Ng D H L,Lu Z Q,et al.J.Alloys Compd.,2005,395:192-200
[8]Wachtler M,Winter M,Besenhard J O.J.Power Sources,2002,105:151-160
[9]Cheng X Q,Shi P F.J.Alloys Compd.,2005,391:241-244
[10]Fang L,Chowdari B V R.J.Power Sources,2001,181:97-98
[11]Wang Y G,Li B,Zhang C L,et al.J.Power Sources,2012,219:89-93
[12]Veeraraghavan B,Paul J,Haran B,et al.J.Power Sources,2002,109:377-387
[13]Park K S,Schougaard S B,Goodenough J B.Adv.Mater.,2007,19(6):848-851
[14]Huggins R A.J.Power Sources,1999,81-82:13-19
[15]Zhang W M,Hu J S,Wan L J,et al.Adv.Mater.,2008,20:1160-1165
[16]Wang J,Too C O,Wallace G G,et al.J.Power Sources,2005,140:162-167
[22]Song J Y,Lee H H,Wang Y Y,et al.J.Power Sources,2002,111:255-267
[23]Zhang Y,Zhang X,Cheng H,et al.Electrochim.Acta,2006,51:4994-5000
[24]Jiang T,Zhang S,Qiu X,et al.Electrochem.Commun.,2007,9(5):930-934
[25]Dimov N,Kugino S,Yoshio M.Electrochim.Acta,2003,48(11):1579-1587
