中國與歐盟質量受權人制度比較及其啟示
2014-07-24 01:41:46陳永法杜一坤
中國藥業 2014年20期
陳永法,杜一坤
(中國藥科大學國際醫藥商學院,江蘇 南京 211198)
質量受權人(QP)這一概念最早由歐盟于1975 年提出。中國從2007 年開始在廣東、安徽、山東等地進行藥品質量受權人試點,2009 年發布的《關于推動藥品生產企業實施藥品質量受權人制度的通知》提出,首先在血液制品類、疫苗類、注射劑類及重點監管特殊藥品類藥品生產企業試行質量受權人制度,2011 年3 月新版《藥品生產質量管理規范》(GMP)正式推行該制度。中國質量受權人制度起步較晚,在實施過程中存在概念模糊、法律地位不明確、支撐體系不完善等問題,筆者現就這幾個方面對歐盟及中國質量受權人制度進行對比分析,并提出完善中國質量受權人制度的合理化建議。
1 對比
1.1 質量受權人的概念及法律地位
中國2010 年版GMP 中明確了質量受權人概念,并將其作為企業生產的關鍵人員。但該部門規章只對其資質、主要職責作了原則性規定,對質量受權人的定位較模糊,且規定質量管理負責人和質量受權人可以兼任。因此,在開展質量受權人制度試點工作中,大部分藥品生產企業的質量受權人由質量管理負責人兼任,出現了“質量負責人”與“質量受權人”共存的現象。目前,法律法規中僅有2010 年版GMP 對質量受權人進行了規定,而《藥品管理法》等上位法并未對質量受權人制度作出要求,僅停留在部門規章層面,缺乏與現行法律法規的有效銜接。
歐盟質量受權人制度的發展較早,其定義質量受權人為由國家藥品監督管理部門認可,負責確保產品按照國家相關法律法規生產、檢驗和放行的人員。該定義強調,質量受權人是經國家藥品監督管理部門審查通過的人員,從業前必須獲得相應的從業資格,且明確界定了其職責。歐盟通過法律和行業守則對質量受權人的職責及行為進行嚴格地規范,如《2003/94/EC指 令》(Directive 2003 /94/EC)、《91/412 /EEC 指 令》(Directive 91/412/EEC)、《2001 /20/EC 指 令》(Directive 2001/20 /EC)、《2001 /82 /EC 指 令》(Directive 2001/82/EC)、《2001 /83 /EC 指令》(Directive 2001 /83 /EC)及《指令文件第4 卷—— 藥品生產企業質量管理規范》(Eudralex Volume 4 - Good Manufacturing Practices)等法律[1],以及《質量受權人申請人及贊助商指導守則》(Qualified Person in the Pharmaceutical Industry Guidance Notes For Applicants and Sponsors)、《質量受權人學習指南》(Qualified Person in the Pharmaceutical Industry Study Guide)、《質量受權人執業守則》(Qualified Person in the Pharmaceutical Industry Code of Practice)等行業守則文件。指令在歐盟具有最高的法律效力,通過不斷更新與完善指令,逐步明確質量受權人在人用藥品生產質量管理、獸用藥生產質量管理、人用藥品臨床試驗管理中應承擔的責任。《質量受權人申請人及贊助商指導守則》[2]主要是對質量受權人5種注冊類型的申請資質、申請材料、申請過程進行詳細分析;《質量受權人執業守則》[3]主要對質量受權人職責進行闡釋;《質量受權人學習指導》[4]對質量受權人的學科背景及實踐經歷進行進一步的要求。行業守則是對指令的進一步細化,為保證質量受權人達到法律要求、切實履行產品質量放行責任起到了很好的保障作用。
1.2 質量受權人申請
目前,中國質量受權人主要是由企業法人從本企業符合受權人資質的人中任命,簽訂授權書后交由省級藥監部門備案,屬于藥品生產企業內部任命程序,沒有標準的操作流程,缺乏規范的選拔、考核標準。在實際生產過程中,部分藥品生產企業對質量受權人的作用未給予足夠重視,為應付當地藥監部門審查而被動設置質量受權人崗位,且質量負責人崗位和受權人崗位的權責交叉,導致質量受權人未很好地與企業原有質量管理體系結合起來,未起到進一步強化和提升藥品質量管理的作用。
與中國最大的不同在于,歐盟質量受權人是一種從業資格,需由申請人提出申請,由衛生部門進行審查并注冊,方能從事質量放行工作。在歐盟,質量受權人有5 種注冊類別:只滿足《2001 /83 /EC 指令》對質量受權人的要求;滿足《2001/83/EC 指令》及《2001/82 /EC 指令》對質量受權人的要求;獸用藥質量受權人;研究用藥品質量受權人;草本藥物質量受權人。前2 種注冊類別,既可成為人用藥品也可成為獸用藥品的質量受權人。
英國衛生部藥品和保健食品監管署(MHRA)及獸藥理事會(VMD)根據《2001 /83 /EC 指令》及《2001 /82 /EC 指令》的要求,將質量受權人評估工作授權給生物學學會(Society of Biology)、英國皇家藥學會(Royal Pharmaceutical Society)及皇家化學會(Royal Society of Chemistry)3 個組織進行。申請人必須成為上述3 個授權組織之一的會員,方可進行申請。其申請前提見圖1,申請過程見圖2。具體申請步驟如下。
填寫申請材料:包括姓名及聯系方式,會員關系,申請類別,相關工作經歷,工作情況,教育經歷,基礎知識(包括藥品相關法律法規及管理、質量受權人工作經歷、質量控制體系),其他知識要求(包括數學及數理統計學、藥物化學及藥理學、藥物制劑及加工、制藥微生物學、分析及測試、藥品包裝、活性藥物成分、研究用藥品),贊助商,聲明,申請次數等;不是初次申請的,還需上交之前所有的申請表格,贊助商報告及面試評價表格等資料。贊助商報告是申請材料的重要組成部分,是贊助商證明申請人能夠滿足《2001 /83 /EC 指令》的相關要求,具有相應的資質且能完成質量受權人職能的證明材料,也是對申請人專業能力的評價報告。
形式審查:一般由2 名來自授權機構的審評員從2 個層面對申請人的申請條件進行審查。一是法律、倫理學及專業課程的學科背景,《2001/83/EC 指令》第49 條[5]闡述了對質量受權人學科背景的總體要求,即申請人應獲得由大學頒發的藥劑學、藥學、獸藥、化學、藥物化學或生物學文憑、證書,或持有其他成員國承認的同等課程學習證書,這些課程包括物理實驗、基礎無機化學、有機化學、藥物化學、藥物分析學、基礎應用生物化學(醫學方向)、生理學、微生物學、藥理學、制藥工藝學、毒理學、生藥學(動植物器官活性物質的組成及作用方向)。二是工作經驗,申請人應從事藥品分析、活性物質檢測及藥品質量檢測工作至少2 年以上;如果大學課程學習時間長達5 年以上,工作經歷可減少1 年;如果理論課學習長達6 年,則工作時間可減少一年半。申請人符合所有條件后進入審評環節。
審評:審評人員對申請人的優、缺點進行客觀、公正地記述。如果申請人在某一方面表現不足,審評人員需提出補充材料,并報質量受權人辦公室審核;如果申請材料符合條件,則進入面試環節。
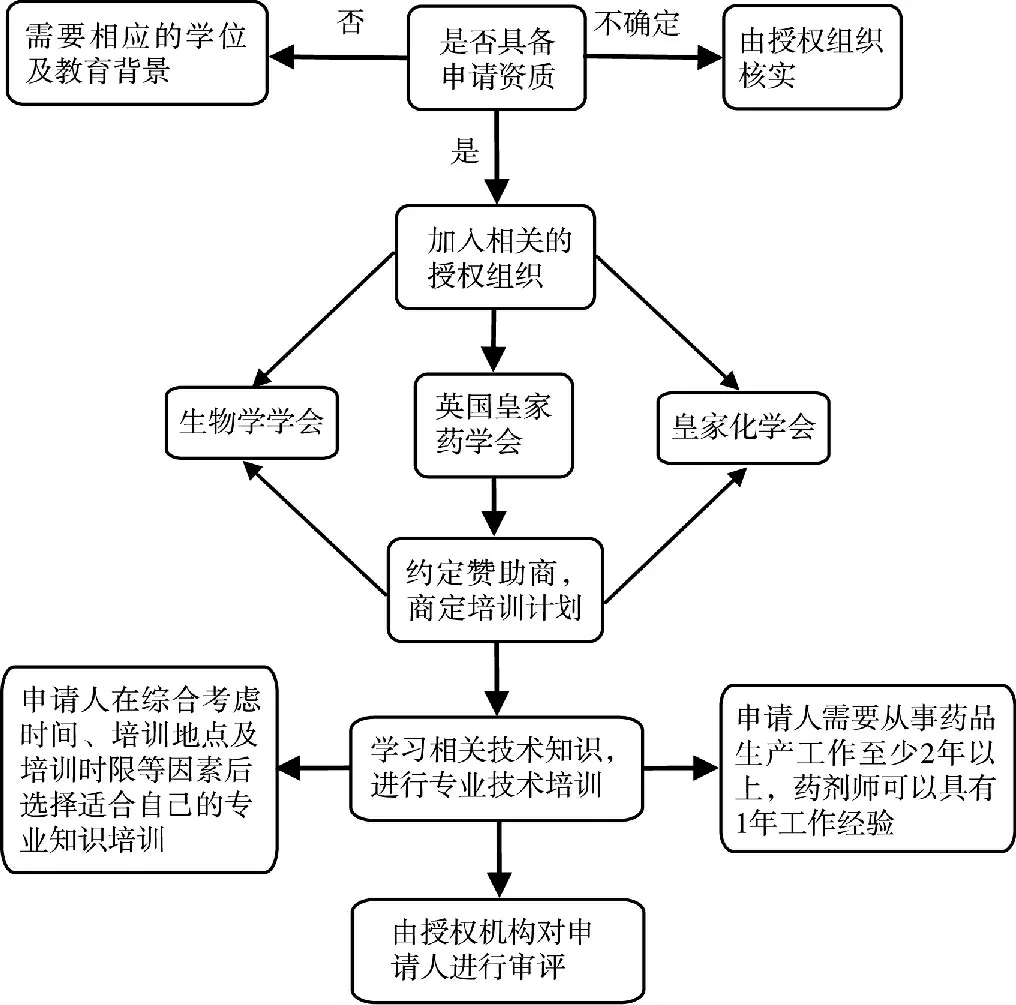
圖1 英國質量受權人申請前提
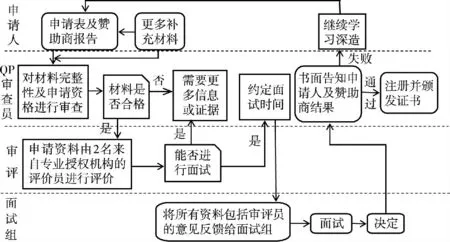
圖2 英國質量受權人申請過程
面試:面試前1 個月,申請人所有申請材料都將送到3 名面試官手中,面試官對材料進行評價,3 名面試官可以隨時溝通,對將在面試中提出的問題達成共識。面試將由來自受權機構的主席主持,面試官將就申請人的專業知識及法律法規進行提問。面試結束后,申請人將收到面試結果通知書,通過面試的申請人將被授予質量受權人證書并進行注冊。沒有通過的,質量受權人辦公室會對申請人的薄弱環節進行總結,并建議再申請的時間。整個申請過程將持續2 ~6 個月。通過審評后,申請人尚不能開展質量受權人的工作,還需要與上市許可持有人簽約,藥品上市許可持有人將申請人的名字填寫到生產許可證上,向MHRA 申請聘任合格的申請人成為質量受權人,MHRA 對申請進行審查并最終作出決定。
1.3 質量受權人支撐體系
1.3.1 就業方式
中國的質量受權人大部分由企業質量負責人兼任,在就業上屬于內部任命的形式,流動性較差;且質量受權人屬于企業員工,受企業法人的領導,個人利益與企業效益之間存在必然聯系,質量受權人的獨立性受到干擾,內部任命制使質量受權人的監督放行職能在實際操作中難以實現。
歐盟的質量受權人是通過注冊認證的方式上崗,在工作地點的選擇上更具有流動性。一般來說,申請人經審查通過后,即與藥品上市許可持有人簽訂聘用合同,并由上市許可持有人向藥品監管部門申請,通過后即成為真正的質量受權人。這種獨具特色的自由簽訂聘用協議的方式可以促進質量受權人在整個歐盟范圍內流通,促使質量受權人資源配置合理化。為幫助質量受權人就業,歐盟質量受權人協會等組織為其提供就業數據庫。數據庫包含大量藥品上市許可持有人的信息,以及質量受權人的基本情況,雙方通過登錄數據庫,就可了解到目前的行業招聘動態。質量受權人可通過數據庫向招聘方發送求職意圖及個人簡歷,如果雙方達成協議,即可與招聘方簽訂聘用合同。同時,通過競聘、審批上崗的擇業形式,也可促使質量受權人不斷完善個人專業技能,進一步激勵質量受權人本身專業素質的提高。
1.3.2 素質培訓
中國2010 年版GMP 第三章第三節規定:“與藥品生產、質量有關的所有人員都應當經過培訓,培訓的內容應當與崗位的要求相適應。除進行本規范理論和實踐的培訓外,還應當有相關法規、相應崗位的職責、技能的培訓,并定期評估培訓的實際效果。”雖然GMP 中對質量受權人素質培養有明確規定,但因無相應配套措施及方案,在制度實施過程中未得到廣泛推廣,可操作性較差。
歐盟比較重視對質量受權人進行素質培養和提升,并在GMP 附錄十六中[6]作出明確規定:質量受權人應該及時更新自己的知識儲備,豐富工作經驗。這就要求質量受權人對藥品生產質量管理體系、GMP 標準、藥品生產及控制以及工作程序有持續的了解。歐盟質量受權人的半開放管理催生了質量受權人培訓產業的蓬勃發展,質量受權人論壇等各種形式的培訓課程應運而生。質量受權人素質培訓包括兩部分:一是申請前的專業課程學習,以使申請人能夠符合申請條件,具備申請資質。如英國科學服務協會為申請人提供一系列的專業素質培訓,主要包括藥品相關法律法規、數學與數據統計、質量受權人角色及職責、藥物化學及治療學、質量控制體系、藥品制劑及制造學、制藥微生物學、藥物活性物質、分析及測試學、研究用藥物、包裝學、生物技術等課程,以保證申請人符合《2001/83 /EC 指令》對申請資質的要求。二是質量受權人的培訓交流,歐盟質量受權人協會定期舉辦交流研討會,方便質量受權人交流在實際工作中遇到的困難及問題,為從業者提升素質創造學習平臺。
2 建議
2.1 確立質量受權人制度的法律地位,完善法律法規體系
建議修訂《藥品管理法》等上位法,明確質量受權人的法律地位與法律責任,保障質量受權人制度的有效推行。同時,各省可通過出臺詳細的規范性文件、配套政策及行業規范等手段,對質量受權人的職責、資質等進行明確規定,使其更具操作性。在提高質量受權人管理辦法法律效力的基礎上,應對其在藥品質量方面所負有的法律責任進行闡述,明確出現質量問題的相關法律責任在企業、企業法人代表、質量受權人之間的追究方式及過錯推定原則,理順《民法通則》規定的企業法人法律責任及質量受權人最終責任的關系[7]。
2.2 逐步推行質量受權人審批制度,引入第三方機制
質量受權人審批制度是指申請人需要通過向省級藥品監督管理部門進行申請,由相關部門負責“質量受權人資格”申請人的審核、注冊、復審工作,對申請人的專業背景、工作經歷及專業技能進行考核,通過審批后,頒發質量受權人從業資格證書,在藥品監督管理部門進行備案管理。審批制度可以提高對質量受權人專業技能及專業能力的考察,提高行業準入門檻,最大限度地保證公平、公正。具體的準入操作可參考英國的模式,采用先核準申請人的基本條件(包括藥學相關專業的學歷及工作經驗要求),合格后接受該專業團體或其他符合資質培訓機構的培訓(培訓課程的內容、安排及培訓質量控制由專業團體統一標準并負責管理),培訓課程結束后經該專業團體考核合格,并在藥品監督管理部門注冊,給予其相應的職業資格。同時,建議引入第三方機制,參考職業經理人經驗,將質量受權人制度發展成為獨立于企業和監督部門的第三方[8]。此舉可進一步提供質量受權人的獨立性和權威性,提供質量受權人職業發展活力,進一步推動該制度的有效實施。
2.3 建立質量受權人支撐體系,保護質量受權人利益
為保障質量受權人利益,建議成立全國性、學術性、公益性的非法人社會組織,如質量受權人專業委員會,由藥品生產質量受權人及藥品安全監管人員自愿參加,搭建質量受權人學習、交流、溝通的平臺。專業委員會的主要職能:開展藥品生產質量管理工作交流;舉辦藥品生產質量管理學術講座;學習研討藥品生產質量管理新的法規要求;優秀受權人的評選、推薦;發行會刊;協助監管部門推動受權人制度實施,完成監管部門交辦任務;舉辦受權人的交流活動等。同時,可借鑒歐盟的經驗,建立質量受權人支撐體系,加強對質量受權人的素質培訓,提升質量受權人的專業素質。由藥品監督管理部門或質量受權人專委會制訂和推行《質量受權人行為準則》或《質量受權人實施指南》,建立從業人員誠信記錄,并對違反該行為準則的質量受權人進行相關處罰,如暫時吊銷或永久取消其職業資格,以加強行業自律[9]。
[1] NSF DBA,The Role and Duties of the EU Qualified Person(QP)[Z].
[2] Society of Biology.Qualified Person in the Pharmaceutical Industry Guidance Notes For Applicants and Sponsors[Z].2013.
[3] Society of Biology.Qualified Person in the Pharmaceutical Industry Code of Practice[Z].2009.
[4] Society of Biology. Qualified Person in the Pharmaceutical Industry Study Guide[Z].2013.
[5] Directive 2001 /83 /EC of the European Parliament and of the council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use[Z].2004.
[6] EMA.Volume 4 Good manufacturing practice(GMP)Guidelines[Z].2001.
[7] 趙明月. 比較制度分析視角下我國藥品質量受權人制度研究[D].哈爾濱:黑龍江中醫藥大學,2012.
[8] 李 鳴. 藥品生產企業質量受權人制度構建研究[D]. 上海:復旦大學,2012.
[9] 高鵬來. 關于完善我國藥品質量受權人制度的研究[D]. 沈陽:沈陽藥科大學,2009.
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
辦公室業務(2020年18期)2020-09-29 12:15:58
遼金歷史與考古(2019年0期)2020-01-06 07:44:44
勞動保護(2019年7期)2019-08-27 00:41:26
中國衛生(2016年7期)2016-11-13 01:06:26
中國衛生(2016年11期)2016-11-12 13:29:18
中國衛生(2016年9期)2016-11-12 13:27:58
中國衛生(2016年5期)2016-11-12 13:25:28
中國衛生(2015年5期)2015-11-08 12:09:48
中國衛生(2014年7期)2014-11-10 02:33:02
