醛固酮與慢性肺源性心臟病肺動脈高壓的相關性研究
2014-08-30 06:51:18覃淑娟陳昌枝
中華肺部疾病雜志(電子版) 2014年4期
關鍵詞:心功能
覃淑娟 陳昌枝
慢性阻塞性肺疾病(chronic obstructive pulmonary disease, COPD)主要的病理基礎是氣道、肺實質、肺血管的慢性非特異性炎癥,炎癥損傷和修復導致氣道和血管壁重塑,從而引起進行性氣流受限及慢性缺氧,其后期往往出現肺動脈高壓而導致慢性肺源性心臟病發生[1]。肺動脈高壓是COPD的重要合并癥,其病理特征為肺動脈平滑肌細胞增殖導致肺動脈血管重塑,其確切發病機制尚未明確[2]。本研究目的旨在探討腎素-血管緊張素-醛固酮系統(renin-angiotensin-aldosterone system, RAAS)中的醛固酮是否參與了肺心病肺動脈高壓的形成。
資料與方法
一、臨床資料
選擇2012年1月至2013年12月我院呼吸內科收治的COPD患者90例,均符合2007 年中華醫學會關于COPD的診斷標準[3],肺功能具有不完全可逆的氣流受限,吸入支氣管擴張劑后,1 s用力呼氣量(forced expiratory volume in one second, FEV1)<80%預計值,FEV1/FVC<70%。根據COPD患者病情分為COPD并肺動脈高壓組,肺心病肺心功能代償期組,慢性肺源性心臟病肺心功能失代償期組。
COPD并肺動脈高壓組:30例患者均符合2004年美國胸科醫師學會(ACCP)及歐洲心臟病學會(ESC)制定肺動脈高壓診斷標準[4-6],及超聲心動圖檢查提示肺動脈高壓的標準[7],但無右心室肥大表現。其中男20 例,女10 例,平均年齡為( 67.82±9.02) 歲。
慢性肺源性心臟病肺心功能代償期組:30例患者為確診COPD 10年以上,病程遷延并發展,符合1977年我國修定的 “慢性肺源性心臟病診斷標準”,同時符合衛生部統編第七版內科學教材慢性肺源性心臟病肺心功能代償期的診斷標準。其中男23例,女7例,平均年齡為(69.38±8.75) 歲。
慢性肺源性心臟病肺心功能失代償期組:30例患者為確診COPD 10年以上,病程遷延并發展,符合1977年我國修定的 “慢性肺源性心臟病診斷標準”,同時符合衛生部統編第七版內科學教材慢性肺源性心臟病肺心功能失代償期的診斷標準。其中男22例 ,女8例,平均年齡為(70±5.65)歲。
所有3組研究對象均經詢問病史、體格檢查、相關實驗室檢查及輔助檢查等排除先天性心臟病、風濕性心臟病、原發性心肌病、冠狀動脈粥樣硬化性心臟病、肺血管疾病、支氣管擴張、肺血管栓塞、支氣管哮喘、原發性及繼發性高血壓、糖尿病、結締組織疾病及其它可能影響心功能、肺動脈壓、肺通氣及換氣功能的疾病。所有3組研究對象在檢查前均通過醫院倫理委員會討論并被告知相關檢查的目的與檢查方法,且均簽署知情同意書,自愿接受檢查。
二、儀器及試劑
主要儀器:意大利COSMED Quark PFT3肺功能儀,PHILIPSIE35心臟B超機,德國西門子血氣分析儀,DFM-96型多管放射免疫計數器,醛固酮定量檢測試劑盒比利時RADIM公司提供。日本三洋電氣公司MCE-192冰箱,北京京立離心機有限公司生產的LD5-2A型臺式低速離心機。
三、研究方法
1. 醛固酮檢測:所有對象于清晨空腹臥位采肘靜脈血,將3 ml血立即注入含有EDTA的試管, 以離心半徑8 cm,3000 r/min 離心10 min,分離血漿,置-20 ℃低溫冰箱保存,按照試劑盒操作說明,采用放射免疫免疫分析法,將待測樣品中的醛固酮和125I-醛固酮與有限的ALD 抗體在適宜條件下進行競爭性結合反應。125I-醛固酮與抗體結合的比例決定于待測樣品或標準品中非標記的醛固酮的含量。抗原抗體復合物用分離試劑沉淀下來,采用DFM-96型多管放射免疫γ計數器測定復合沉淀物的放射性計數。
2. 心臟彩超檢查:均由一名經驗豐富的副主任醫師使用彩色多普勒超聲心動圖儀操作。用連續波Doppler法(CW)根據返流壓差估測肺動脈壓[8]。取三尖瓣返流頻譜,選擇心尖四腔心切面,取樣容積置于三尖瓣返流束起始處,取樣線方向同樣盡量平行于返流束方向,測得三尖瓣返流最高流速,根據簡化Bernoulli方程(ΔP=4V2,V為最大返流速度) 可求得右心室與右心房之間壓差。若無右室流出道梗阻,肺動脈收縮壓(sPAP)與右心室收縮壓(sRVP)相似,即: sPAP=sRVP=右房壓(RAP) +三尖瓣跨瓣壓差(ΔP)。通過二維超聲心動圖測量左房右房橫徑,右室舒張末內徑(RVEDD)、左室舒張末內徑(LVEDD),右心室流出道內徑、右心室內徑、右心室前壁厚度等。
3. 動脈血氣分析:在未予吸氧的條件下,取橈動脈血2 ml, 用德國西門子全自動血氣分析儀測定動脈血氧分壓( partial pressure of oxygen, PaO2)、 血二氧化碳分壓( partial pressure of carbon dioxide, PaCO2)等血氣指標測定。
四、統計學方法
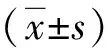
結 果
一、臨床資料比較
比較各組間的臨床資料,年齡、體重指數、吸煙指數差異無統計學意義(P>0.05),在肺功能FEV1占預計值比較上,慢性肺源性心臟病失代償期組的FEV1占預計值低于COPD并肺動脈高壓組、慢性肺源性心臟病代償期組,差異有統計學意義(P<0.05),見表1。
二、患者血漿醛固酮濃度、肺動脈壓力、PaO2、PaCO2
3組患者血漿醛固酮濃度、肺動脈壓力、PaO2、PaCO2比較,差異均有統計學意義(P<0.05),其中慢性肺源性心臟病失代償期組的醛固酮水平、肺動脈壓、二氧化碳分壓均高于COPD并肺動脈高壓組、慢性肺源性心臟病代償期組,差異有統計學意義(P<0.05)。慢性肺源性心臟病失代償期組的動脈血氧分壓水平低于COPD并肺動脈高壓組、慢性肺源性心臟病代償期組,見表2。
三、血漿醛固酮水平與肺動脈壓力、PaO2、PaCO2和FEV1占預計值的相關性
血漿醛固酮水平與肺動脈壓力成線性正相關(r=0.514,P=0.001),與血氧分壓呈負相關(r=-0.248,P=0.019),差異均有統計學意義。血漿醛固酮水平與二氧化碳分壓、FEV1%無相關(分別為r=0.155P=0.146,r=-0.078P=0.464),差異無統計學意義。
討 論
醛固酮是RAAS系統中的重要組成部分,是最強的鹽皮質激素。目前的研究發現,醛固酮對心血管系統的影響除了引起潴鈉排鉀之外,還具有引起心肌纖維化、血管重塑、心室肥大等作用。肺動脈高壓為COPD的常見并發癥,表現為內皮細胞功能障礙和平滑肌細胞的異常增生,導致肺血管的異常收縮和遠端肺小動脈的重構,進而出現中膜肥厚內膜增生叢狀損害和血栓形成等病理改變,其病理特征為肺動脈平滑肌細胞的增殖導致肺動脈血管重塑[9]。因此,具有致心肌纖維化、血管重塑、心室肥大等作用的醛固酮可能參與了慢性肺源性心臟病肺動脈高壓的形成。
本研究結果顯示,慢性肺源性心臟病失代償期組的醛固酮水平、肺動脈壓水平均高于COPD并肺動脈高壓組、慢性肺源性心臟病代償期組,差異有統計學意義(P<0.05)。其原因可能為慢性肺源性心臟病失代償期患者由于右心功能不全,腎素-血管緊張素-醛固酮系統被激活,導致醛固酮分泌增多,而嚴重右心功能不全可累及左心功能,使左房壓增高,又可出現被動性肺動脈高壓,從而導致肺動脈高壓的嚴重程度增加。另外本研究也發現肺動脈壓力隨著醛固酮濃度的增加而升高,兩者呈顯著的正相關關系,這與徐紅蕾等[10]報告一致,表明醛固酮與慢性肺源性心臟病患者的肺動脈高壓有一定的相關關系。而肺動脈壓力隨著動脈血氧分壓的降低而升高,兩者呈低度負相關關系,表明缺氧亦參與了肺動脈高壓的形成。
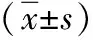
表1 3組間臨床資料比較
注:與COPD并肺動脈高壓組、慢性肺源性心臟病代償期組比:aP<0.05
表2 3組間醛固酮濃度、肺動脈壓、PaO2、PCO2比較
注:與COPD并肺動脈高壓組、慢性肺源性心臟病代償期組比:aP<0.05(1 mmHg=0.133 kPa)
COPD 引起的肺動脈高壓屬于一種多因性肺動脈高壓,本研究結果顯示醛固酮與肺肺動脈壓力呈顯著的正相關,血醛固酮濃度增加可致肺動脈高壓進一步惡化,右心功能不全加重。其作用機制可解釋為:①醛固酮通過激活鹽皮質激素受體,可導致血管重塑與靶器官損傷[11];②醛固酮可刺激肺動脈平滑肌細胞增生致血管重塑,這種作用可被醛固酮受體拮抗劑依普利酮所抑制[12];③增多的醛固酮通過降低血管抗氧化能力導致血管內皮功能障礙,并損害血管反應性,增強氧化應激,并限制一氧化氮的生物活性[13];④增多的醛固酮激活交感神經系統,降低了壓力感受器敏感性,增加電解質排泄量(K+,Mg2+),并參與細胞凋亡等[14];⑤醛固酮可激活炎癥反應,通過增加纖溶酶原激活物抑制劑-1的表達和促進組織纖維化[15]。
肺動脈壓的高低,直接影響到COPD患者的預后,如能早期有效地控制肺動脈高壓,對于提高COPD患者的生存質量,改善預后,防止慢性肺源性心臟病的發生發展具有重要社會及經濟效益。而目前COPD肺動脈高壓形成機制復雜,涉及到基因、細胞、體液、分子等多因素的綜合作用,其具體機制目前尚不完全清楚,因此醛固酮在COPD 肺動脈高壓形成中的作用機制還有待進一步探討。
參 考 文 獻
1 董佳慧, 孫耕耘. 結締組織病相關肺動脈高壓的研究進展[J/CD]. 中華肺部疾病雜志: 電子版, 2014, 7(1): 104-107.
2 曾天星, 洪旭初. 慢性阻塞性肺疾病的表型及治療[J/CD]. 中華肺部疾病雜志: 電子版, 2014, 7(2): 216-219.
3 中華醫學會呼吸病學分會慢性阻塞性肺疾病學組. 慢性阻塞性肺疾病(COPD)診治指南(2007年修訂版)[J]. 中華結核和呼吸雜志, 2007, 30(1): 8-17.
4 Proceeding of 3nd World symposium on pulmonary arterial hypertension.Venice, Italy, June 23-25, 2003[J]. J Am Coll Cardiol, 2004, 43(2 Suppl S): 1S-90S.
5 Diagnosis and management of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines[J]. Chest, 2004, 126(Suppl 1): 7S-10S.
6 Galiè N, Torbicki A, Barst A, et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The task force on diagnosis and treatment of pulmonary arterial hypertension of the european society of cardiology[J]. Eur Heart J, 2004, 25(24): 2243-2278.
7 McQuillan BM, Picard MH, Leavitt M, et al. Clinical correlates and reference intervals for pulmonary artery systolic pressure among echocardiographically normal subjects[J]. Circulation, 2001, 104(23): 2797-2802.
8 Berger M, Haimowitz A, Van Tosh A, et al. Quantitative assessment of pulmonary hypertension in patients with tricuspid regurgitation using continuous wave Doppler ultrasound[J]. J Am Coll Cardiol, 1985, 6(2): 359-365.
9 Bradford CN, Ely DR, Raizada MK. Targeting the vasoprotective axis of the renin-angiotensin system: a novel strategic approach to pulmonary hypertensive therapy [J]. Curr Hypertens Rep, 2010, 12(4): 212-219.
10 徐紅蕾, 陳少賢, 蔡映云, 等. 醛固酮與肺心病患者肺動脈高壓的關系[J]. 溫州醫學院學報, 2006, 36(4): 353-355.
11 Fiebeler A, Muller DN, Shagdarsuren E, et al. Aldosterone, mineralocorticoid receptors, and vascular inflammation[J] . Curr Opin Nephrol Hypertens, 2007, 16(2): 134-142.
12 Yamanaka R, Otsuka F, Nakamura K, et al. Involvement of the bone morphogenetic protein system in endothelin and aldosterone-induced cell proliferation of pulmonary arterial smooth muscle cells isolated from human patients with pulmonary arterial hypertension[J]. Hypertens Res, 2010, 33(5): 435-445.
13 Leopold JA, Dam A, Maron BA, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphatede hydrogenase activity [J]. Nat Med, 2007, 13(2): 189-197.
14 Carey RM. Aldosterone and cardiovascular disease [J]. Curr Opin Endocrinol Diabetes Obes, 2010, 17(3): 194-198.
15 Gaddam KK, Verma A, Thompson M, et al. Hypertension and cardiac failure in its various forms[J]. Med Clin North Am, 2009, 93(3): 665-680.
猜你喜歡
中華養生保健(2020年8期)2021-01-14 01:13:58
心肺血管病雜志(2020年5期)2021-01-14 00:43:54
天津醫科大學學報(2019年6期)2019-08-13 07:04:34
護士進修雜志(2017年2期)2017-02-26 14:32:29
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:28
中國衛生標準管理(2015年18期)2016-01-20 09:27:12
中國衛生標準管理(2015年13期)2016-01-15 02:58:25
哈爾濱醫藥(2015年2期)2015-12-01 03:57:18
中國當代醫藥(2015年22期)2015-03-01 02:05:31
中國醫藥科學(2015年2期)2015-02-27 12:32:09
