模擬高原環境導致的低碳酸血癥對家兔肺動脈壓力的影響
2014-08-30 06:51:08王超平趙云峰
中華肺部疾病雜志(電子版) 2014年4期
王超平 高 芬 趙云峰
急性高原反應患者存在低氧血癥,動脈血氧分壓降低,刺激頸動脈體和主動脈體的化學感受器,反射性的引起呼吸加深加快,從而使肺泡通氣量代償性的顯著增加,PaCO2降低,出現呼吸性堿中毒。低氧環境,氧與血紅蛋白結合減少,加之釋氧量增加,使血液中偏堿性的脫氧血紅蛋白增多,使血液的pH值增高。同時,由于PaCO2分壓降低,CO2對呼吸中樞的刺激作用減弱,造成睡眠時周期性呼吸和呼吸暫停,導致睡眠時低氧血癥,進一步加重了缺氧[1],從而導致多種高原病的發生。在高原地區由于呼吸增強排出過多CO2,產生低碳酸血癥往往是高原病的發病因素之一。低氧可引起血管內皮細胞損傷,血管內皮合成和分泌的各種血管舒張因子平衡失調,導致早期的肺血管收縮(hypoxic pulmonary vasoconstriction, HPV) 以及后期的肺血管重建(hypoxic pulmonary vascular remodeling, HPSR)。HPSR是低氧性肺動脈高壓的重要病理基礎[2],目前,國內外肺動脈高壓研究均為低氧所致肺動脈高壓,罕見低碳酸血癥對肺循環壓力影響的報道資料,但有文獻報道雌激素對肺動脈壓力有一定的影響[3]。本研究以健康雄性家兔為實驗對象,旨在觀察模擬高原環境過度通氣所致低碳酸血癥對家兔肺動脈壓的影響
材料與方法
一、試劑和材料
試劑和材料: ① 健康雄性清潔級家兔10只,兔齡6~7個月,體重2.2±0.3 kg(種屬為青紫藍 由蘭州大學醫學院實驗動物中心提供); ②SNIBE MAGLUMI 2000 PLUS全自動化學發光免疫分析系統(深圳新產業公司) ;③血管緊張素II(AngII)測定試劑盒(深圳市新產業生物醫學工程有限公司);④BL-820生物機能實驗系統(成都泰萌科技有限公司);⑤HX-300動物呼吸機(成都泰萌科技有限公司);⑥MEDICA血氣分析儀(美國MEDICA公司)。
二、實驗方法
1.動物試驗步驟:所有家兔用20% 烏拉坦3~4 ml/kg行耳緣靜脈穿刺注射麻醉。麻醉成功后將家兔固定于手術臺上,氣管插管,并行頸外靜脈插管至肺動脈,BL-820生物機能實驗系統監測肺動脈壓;所有家兔自主呼吸15 min做為空白對照(A階段);接HX-300動物呼吸機行機械通氣15 min(B階段),氧濃度為21%,呼吸機參數設置(潮氣量5~8 ml/kg,頻率45~55次/min,吸呼比:1︰2)。低碳酸血癥造模呼吸機的參數(潮氣量8~10 ml/kg,頻率75~85次/min,吸呼比:1︰2~2.5),各階段持續時間為20 min。輕度低碳酸血癥(C階段,動脈血氣分析PaCO225~35 mmHg, pH>7.45),中度低碳酸血癥(D階段,血氣分析PaCO215~25 mmHg, pH>7.50),重度低碳酸血癥(E階段,血氣分析PaCO2<15 mmHg, pH>7.55)[4-6]。
2.血液標本采集及血清AngⅡ測定:機械通氣時間達到上述水平后分別抽取家兔耳緣靜脈血,EDTA抗凝。將所采取的兔血標本以離心半徑8 cm,3000 r/min,離心15 min,分離血清,血清存于-80 ℃冰箱保存。血清AngⅡ測定嚴格按照試劑盒說明進行操作。
3.肺動脈平均壓力的測定:麻醉生效后,鈍性分離頸外靜脈,用自制的硅膠管沿頸外靜脈至右心房、右心室、肺動脈。根據波形變化確定插管到達的正確位置,測定肺動脈收縮壓(pulmonary artery systolic pressure, sPAP)和肺動脈舒張壓(diastolic pulmonary artery pressure, dPAP),并計算出肺動脈平均壓(mean pulmonary artery pressure, mPAP)。
三、統計學方法
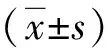
結 果
一、空白對照、無低碳酸血癥、低碳酸血癥各階段動脈血氣結果的比較
結果顯示:①家兔pH、PaCO2、PaO2在A階段至B階段,差異無統計學意義(P均>0.05)。C階段至E階段,隨著低碳酸血癥的加重,pH由(7.47±0.03)升至(7.58±0.03),PaCO2由(30.10±2.88 mmHg)降至(11.40±2.07 mmHg),PaO2由(85.00±4.14 mmHg)升至(93.20±3.49 mmHg),差異均有統計學意義(P均<0.05);②A階段至C階段,家兔MPAP之間的差異無統計學意義(P均>0.05)。C階段至E階段,隨著低碳酸血癥的加重,MPAP由(15.15±0.78 mmHg)降至(12.76±0.42 mmHg),差異有統計學意義(P<0.05)。
二、AngⅡ變化情況A階段至C階段,家兔血清AngⅡ變化不明顯,差異無統計學意義(P>0.05)。C階段至E階段,隨著低碳酸血癥的加重,血清AngⅡ由(83.08±6.74 pg/ml)降至(62.84±6.20 pg/ml),差異有統計學意義(P<0.05),見表1。
討 論
高原肺動脈高壓主要機制為缺氧導致肺血管收縮,血流阻力增大,從而引起肺動脈壓升高。
本實驗表明,輕度低碳酸血癥階段至重度低碳酸血癥階段,隨著機械通氣呼吸頻率的增加和吸呼比的改變,低碳酸血癥進行性加重,pH值由(7.47±0.03)升至(7.58±0.03),PaCO2由(30.1±2.88 mmHg)降至(11.40±2.07 mmHg),PaO2由(85.00±4.14 mmHg)升至(93.20±3.49 mmHg),差異均有統計學意義。在吸入氧濃度不變的情況下,隨PaCO2的下降,PaO2卻在升高,考慮其機制可能為過度通氣能使更多的新鮮空氣進入肺泡,從而提高肺泡腔的PaO2,改善高原環境導致的機體缺氧。通過過度通氣能把原來未參與換氣的肺泡調動起來,以增大呼吸面積,提高氧的彌散。另外,血液堿性環境下血紅蛋白在肺部結合氧能力增加,而在組織中血紅蛋白釋氧相應減少。
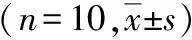
表1 家兔實驗各階段動脈血氣結果比較及AngⅡ變化
注:a與A階段比較,P<0.05 ;b與B階段比較,P<0.05;c與C階段比較,P<0.05;d與D階段比較,P<0.05
本實驗發現輕度低碳酸血癥階段至重度低碳酸血癥階段,隨著低碳酸血癥的加重,mPAP逐漸降低,中度、重度低碳酸血癥能導致肺動脈壓力降低,原因可能與前列腺環素(prostacyclin, PGI2)有關。Nishio等[7]研究人類肺動脈內皮細胞的實驗認為,CO2相關的pH改變影響PGI2的產生,PGI2是由內皮細胞依賴Ca2+濃度而產生的舒張性血管活性類前列腺素,堿中毒時細胞外Ca2+內流增加,PGI2的產生顯著增加,因而肺血管舒張,肺動脈壓力降低。
AngⅡ是腎素-血管緊張素-醛固酮系統(renin-angiotensin-aldosterone system, RAS)中最重要的活性成分,在體內有多種重要的功能,它作用于心臟、血管平滑肌、腎臟和腎上腺等組織的AngⅡ受體,導致血管收縮、醛固酮釋放、水鈉潴留、平滑肌增生等[8]。血液循環內的AngⅡ主要在肺內產生,AngⅠ在肺內血管經張素轉換酶(angiotensin converting enzyme, ACE)作用下形成具有增壓作用更強的AngⅡ。而且肺循環是體內唯一不使AngⅡ滅活的血管床,肺循環流出的血液中AngⅡ的含量最高[9]。當機體處于低氧狀態時,ACE活性增強,引起肺小動脈收縮,形成低氧性肺動脈高壓(hypoxic pulmonary hypertension, HPH),是急、慢性高原病的發生機制之一[10]。
血管局部RAS也產生AngⅡ,并以自分泌方式作用于血管平滑肌細胞或經過旁分泌方式影響內皮細胞的功能。缺氧可促進AngⅡ的釋放,導致肺血管收縮反應增強。AngⅡ除直接作用于肺血管外,還通過刺激腎上腺能神經末梢增加去甲腎上腺素的釋放,間接調節肺血管張力[11]。研究發現在體外實驗中,AngⅡ能引起肺血管平滑肌鈣離子內流和血管收縮[12]。本實驗結果提示,輕度低碳酸血癥階段至重度低碳酸血癥階段,隨著低碳酸血癥的加重,血清AngⅡ由(80.21±7.91 pg/ml)降至(65.84±6.19 pg/ml),差異具有統計學意義。
綜上所述,本實驗模擬高原環境導致的低碳酸血癥,研究不同程度的低碳酸血癥對家兔平均肺動脈壓力及血清AngⅡ的水平的關系。結果顯示:中重度的低碳酸血癥對肺循環的影響較大,使平均肺動脈壓力下降,對高原低氧導致的肺動脈壓力升高可能有一定的拮抗作用,血清AngⅡ水平隨著MPAP的下降而降低,從而可能使高原低氧導致肺血管收縮作用減弱。
參 考 文 獻
1 高鈺琪, 主編. 高原病理生理學[M]. 北京: 人民衛生出版社, 2006: 157-160.
2 崔建華. 缺氧性肺動脈高壓的研究進展[J]. 西北國防醫學雜志, 2013, 34(3): 251-255.
3 孫思慶, 林 勇, 朱曉莉, 等. 雌激素及其受體對低氧性肺血管重建的作用[J]. 中國病理生理雜志, 2007, 23(1): 76-80.
4 Yundt KD, Diringer MN. The use of hyper-ventilation and its impact on cerebral ischemia in the treatment of traumatic brain injury[J]. Crit Care Clin, 1997, 13(1): 163-184.
5 Allen CH, Ward JD. An evidence- based approach to management of increased intracranial pressure [J]. Crit Care Clin, 1998, 14(3): 485-495.
6 Marion DW, Spiegel TP. Changes in the management of severe traumatic brain injury: 1991-1997[J]. Crit Care Med, 2000, 28(1): 16-18.
7 Nishio K, Suzuki Y, Takeshita K, et al. Effects of hypercapnia and hypocapnia on [Ca2+]i mobilization in human pulmonary artery endothelial cells[J]. J Appl Physiol, 2001, 90(6):2094-2100.
8 冉海紅, 張 然. 腎素-血管緊張素系統通過氧化應激機制參與血管反應性的調節[J]. 生理科學進展, 2011, 42(2): 117-120.
9 Veerappan A, Reid AC, Estephan R, et al. Mast cell renin and a local renin angiotensin system in the airway role in Bronchial obstruction[J]. Proc Natl Acad Sci USA, 2008, 105(4): 1315-1320.
10 劉世明, 余滿堂, 吳天一. 青海漢族人群ACE基因多態性頻率的研究[J]. 高原醫學雜志, 2009, 19(3): 30.
11 王 媛, 楊俊玲. 血管緊張素Ⅱ在肺部疾病中的作用[J/CD]. 中華哮喘雜志:電子版, 2013, 7(4): 289-292.
12 Stenmark KR, McMutry IF. Vascular remodeling verus vasoconstriction in chronic hypoxic pulmonary hypertention: A time for veapprasisal? [J]. Circ Res, 2005, 97(2): 95-98.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
中老年保健(2021年3期)2021-08-22 06:50:04
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
