HPLC測定骨松寶顆粒中淫羊藿苷和朝藿定C的含量
2014-09-03 08:45:18穆建國甘惠仍賈憲生
中國合理用藥探索 2014年6期
穆建國 甘惠仍 賈憲生
(1貴州富華藥業(yè)有限責(zé)任公司,貴州 黔南州 551206;2貴陽中醫(yī)學(xué)院,貴陽550002)
骨松寶顆粒是我公司獨家生產(chǎn)的純中藥制劑(批準文號:國藥準字Z52020006),藥物組成為淫羊藿、續(xù)斷、川芎、赤芍、知母、生地黃、莪術(shù)、三棱和牡蠣等,具有補腎活血、強筋壯骨的功效,臨床用于骨瘺(骨質(zhì)疏松)引起的骨折、骨痛、骨關(guān)節(jié)炎,以及預(yù)防更年期骨質(zhì)疏松等癥。骨松寶顆粒處方收載于《衛(wèi)生部藥品標準(中藥成方制劑第十七冊)》(WS-B-3292-98),原質(zhì)量標準未制定含量測定項目,難以滿足對產(chǎn)品質(zhì)量控制的需要。本研究選擇處方中君藥淫羊藿所含較多的藥理活性成分異戊烯基取代的黃酮類化合物淫羊藿苷和朝藿定C[1]作為指標成分,采用高效液相色譜法(HPLC)對骨松寶顆粒中淫羊藿苷和朝藿定C的含量進行測定[2],用以評價主藥淫羊藿含量,該方法簡便、快速、準確,重復(fù)性好,為有效提高骨松寶顆粒質(zhì)量標準控制產(chǎn)品的質(zhì)量提供了依據(jù)。
1 儀器與試藥
1.1 儀器
島津LC-10ATvp高效液相色譜儀(二元泵,SPD-10Avp紫外檢測器,WML-2010色譜工作站);CX-250型超聲波清洗器(功率 300W);AE-240電子分析天平[梅特勒-托利多儀器(上海)有限公司]。
1.2 試藥
對照品:淫羊藿苷(原中國藥品生物制品檢定所提供,含量測定用,批號:110737-200415),朝藿定C(原中國藥品生物制品檢定所提供,含量測定用,批號:110780-200801);甲醇為色譜純;水為高純水,其他試劑為分析純;骨松寶顆粒:貴州富華藥業(yè)有限責(zé)任公司提供(含糖型,批號20110106)。
2 方法與結(jié)果
2.1 對照品溶液的制備
精密稱取對照品朝藿定C和淫羊藿苷,加適量甲醇制成濃度分別為 1.076,0.020 8 mg/mL的溶液,作為對照品溶液。
2.2 供試品溶液的制備
取供試品,混勻,研細,取約2 g,精密稱定,置具塞錐形瓶中,精密加入甲醇20 mL,密塞,稱定重量,超聲處理 1 h,放冷至室溫,再稱定重量,用甲醇補足減失的重量,搖勻,濾過,取續(xù)濾液,即得。
2.3 色譜條件
色譜柱:大連依利特分析儀器有限公司Hypersil ODS2 (250 mm × 4.6 mm,5 μm);柱溫:25 ℃;流動相:乙腈-0.1%磷酸溶液(25∶75)為流動相;流速1 mL/min;檢測波長 270 nm;進樣量:10 μL。
2.4 測定方法
分別精密吸取對照品溶液與供試品溶液各10 μL,注入高效液相色譜儀,測定對照品與供試品峰面積,計算。按上述色譜條件獲得對照品和樣品的色譜圖見圖1,2。
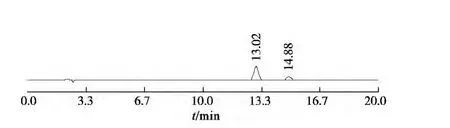
圖1 朝藿定和淫羊藿苷混合對照色譜圖(左為朝藿定C,右為淫羊藿苷)
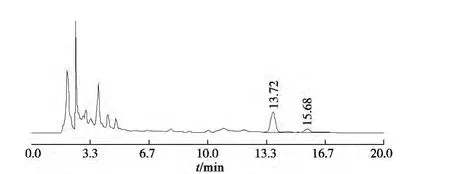
圖2 供試品色譜圖
2.5 線性關(guān)系考察
精密稱取對照品朝藿定C淫羊藿苷,加適量甲醇制成其濃度分別約為 1.076,1.039 mg/mL的溶液,作為單一成分對照品儲備溶液。精密吸取朝藿定的對照品溶液C 2.0 mL和淫羊藿苷的對照品溶液0.4 mL,置10 mL容量瓶中,加甲醇定容至刻度,搖勻,即得到每1 mL含朝藿定C和淫羊藿苷分別為0.215 2和0.041 6 mg的混合對照品溶液,精密吸取混合對照液 1,2,4,5,8,10,15,20 μL分別進樣,按上述色譜條件測定,記錄峰面積。以峰面積積分值(Y)為縱坐標、朝藿定C和淫羊藿苷對照品的量(X)為橫坐標,繪制標準曲線。得朝藿定C近似過原點的線性回歸方程:
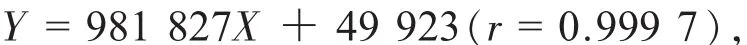
線性范圍為 0.216~ 4.303 μg;得淫羊藿苷近似過原點的線性回歸方程:
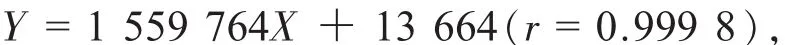
線性范圍為0.042~ 0.831 μg。線性測定結(jié)果見表1。
2.6 精密度試驗
取骨松寶顆粒劑適量混勻,研細,取2 g,精密稱定,按含量測定項下方法制備供試液,連續(xù)進樣9次,測得朝藿定C峰面積平均值1 431 761,RSD=0.13%,淫羊藿苷峰面積平均值260 823,RSD=0.99%,表明精密度良好。
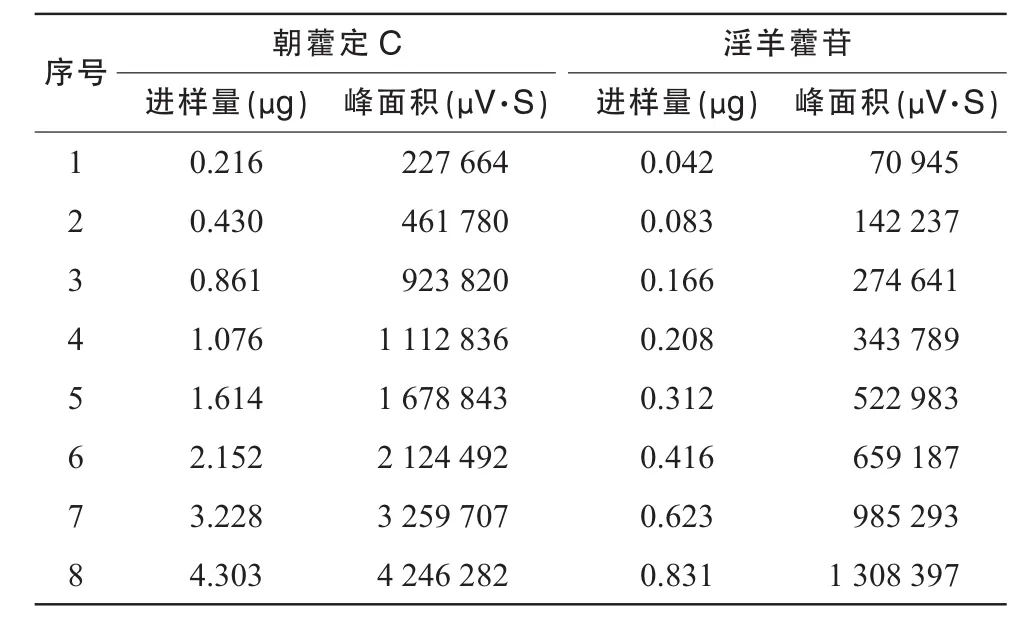
表1 朝藿定C和淫羊藿苷線性測定結(jié)果
2.7 重復(fù)性試驗
取骨松寶顆粒適量混勻,研細,分別取6份,各2 g,精密稱定,按質(zhì)量標準草案含量測定項制備供試液,精密吸取供試液進樣,計算朝藿定C含量和淫羊藿苷含量,朝藿定C含量平均值為1.477 mg/g,RSD=1.64%;淫羊藿苷含量平均值為0.166 mg/g,RSD=1.73%,表明重復(fù)性良好。
2.8 穩(wěn)定性試驗
取骨松寶顆粒適量混勻,研細,取2 g,精密稱定,按含量測定項下方法制備供試液,精密吸取10 μL,在時間間隔為 0,2.0,4.0,6.0,8.0,10.0 h,分別進樣供試品朝藿定C峰面積平均值1 337 801,RSD=1.98%,淫羊藿苷峰面積平均值 248 486,RSD=1.89%,結(jié)果表明本試驗方法在10 h內(nèi)穩(wěn)定。
2.9 回收率試驗
取骨松寶顆粒適量,混勻,研細,分別取9份,各1 g,精密稱定,于1,2,3號樣中分別加入朝藿定C對照品溶液(C=1.076 mg/mL)0.7 mL和淫羊藿苷對照品溶液(C=0.103 9 mg/mL)0.7 mL,相當(dāng)于0.753 2 mg朝藿定C對照品和0.072 7 mg淫羊藿苷對照品;于4,5,6號樣中分別加入朝藿定C對照品溶液(C=1.076 mg/mL)1.4 mL和淫羊藿苷對照品溶液(C=0.103 9 mg/mL)1.5 mL,相當(dāng)于1.506 4 mg朝藿定C對照品和0.155 9 mg淫羊藿苷對照品;于7,8,9號樣中分別加入朝藿定C對照品溶液(C=1.076 mg/mL)2.1 mL和淫羊藿苷對照品溶液(C=0.103 9 mg/mL)2.2 mL,相當(dāng)于2.259 6 mg朝藿定C對照品和0.228 6 g淫羊藿苷對照品。按質(zhì)量標準草案含量測定項制備供試液,進樣,計算朝藿定C和淫羊藿苷回收率,朝藿定C回收率為98.53%~ 101.92%,RSD=0.98%,淫羊藿苷97.32%~103.69%,RSD=1.98%,見表2。
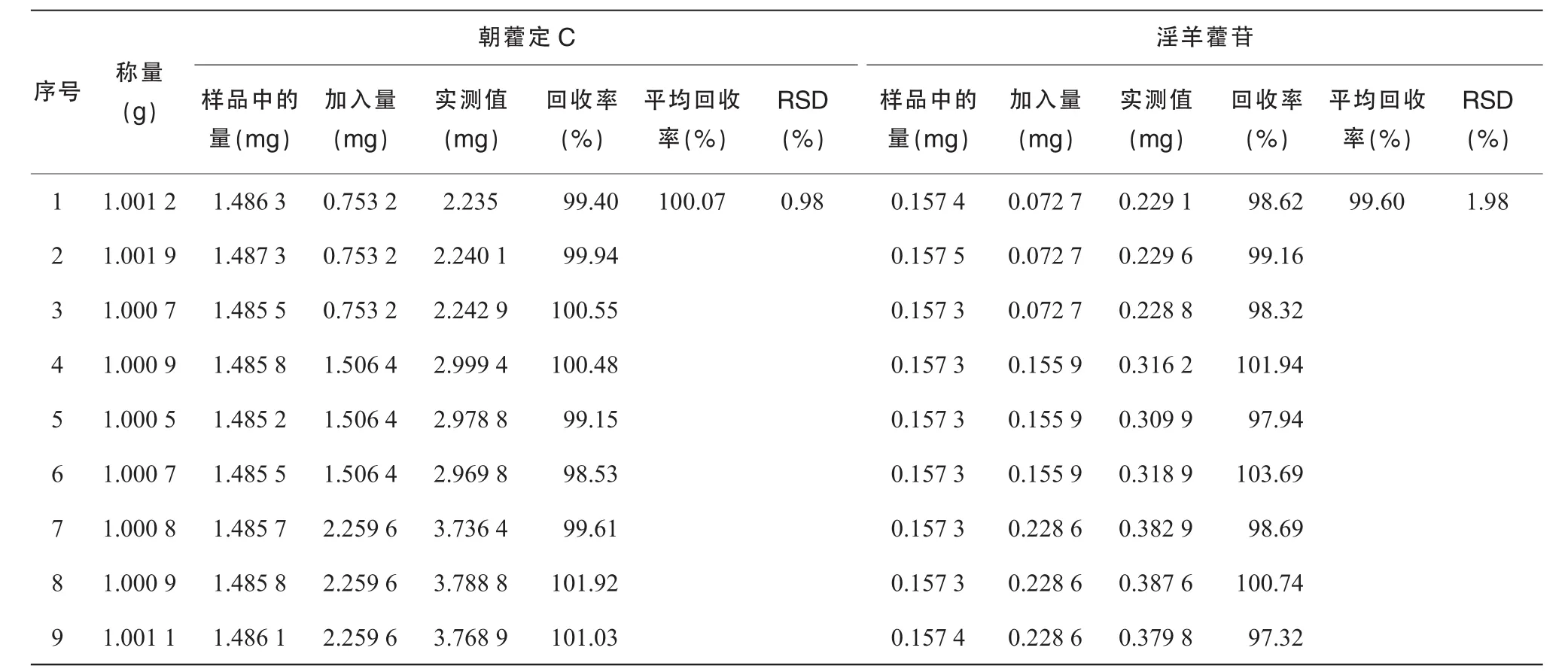
表2 加樣回收試驗測定結(jié)果(n=9)
2.10 樣品含量測定
取10批骨松寶顆粒(含糖型),按2.2項下方法制備供試品溶液,按上述色譜條件測定,計算含量,結(jié)果見表3。

表3 10批骨松寶顆粒中朝霍定C和淫羊藿苷的含量測定結(jié)果 (mg/g)
3 討論
3.1 檢測波長的確定
骨松寶顆粒中含量測定的指標成分朝藿定C和淫羊藿苷為黃酮類成分,在265~ 275 nm和340~350 nm均有吸收。經(jīng)對本品色譜分析檢測波長 220,260,270,340 nm 的考察,在 270 nm 波長處朝藿定C和淫羊藿苷吸收峰靈敏度較高,其他色譜峰干擾相對較小,色譜峰的分離最為理想,故選用270 nm作為檢測波長。
3.2 色譜柱的選擇
分別考察了EliteHypersilODS2(5μm,250mm×4.6 mm)色譜柱和 Diamonsil(鉆石)C18(5 μm,250 mm ×4.6mm)色譜柱,結(jié)果兩個廠牌均可,Diamonsil色譜柱出峰時間較長,故選擇前者。
3.3 流動相的選擇
考察了甲醇-1%冰醋酸,乙腈-水,乙腈-0.1%磷酸等組成的流動相分離情況,以乙腈-0.1%磷酸系統(tǒng)較好;又考察了不同比例乙腈-0.1%磷酸(24∶76,25∶75,26∶74,28∶72)為流動相的分離情況,以乙腈-0.1%磷酸比例為 25∶75和 26∶74對兩個成分的綜合分離效果較好,后者出峰時間稍短,故確定以前者乙腈-0.1%磷酸(25∶75)為流動相比例;流速考察了 1.2,1.0,0.8 mL/min,除柱壓和保留時間有較大差異外分離結(jié)果區(qū)別不大,1.0和 0.8 mL/min流速分離較1.2 mL/min流速分離稍好,故確定為1 mL/min。
3.4 指標性成分的確定
淫羊藿作為處方中君藥,其主要活性成分為黃酮類化合物[3-4],其中朝藿定C含量較高[5],《中國藥典》(2010年版)除巫山淫羊藿以朝藿定C外其余品種僅以淫羊藿苷作為藥材質(zhì)量控制標準[6],不能客觀地反映和評價淫羊藿藥材的內(nèi)在質(zhì)量[7],故考慮選擇淫羊藿苷和朝霍定C作為質(zhì)量控制指標。
本方法簡便、快速、準確,重復(fù)性好,能較全面地控制骨松寶顆粒的質(zhì)量,在實際使用中,為便于評價,可以將骨松寶顆粒中朝藿定C和淫羊藿苷的含量之和用以衡量制劑中淫羊藿含量進而作為骨松寶顆粒含量的質(zhì)量控制指標。本研究結(jié)果為骨松寶顆粒質(zhì)量標準的提升提供了依據(jù)。
[1]孟寧,孔凱,李師翁.淫羊藿屬植物化學(xué)成分及藥理活性研究進展[J].西北植物學(xué)報,2010,30(5):1063-1073.
[2]李曉龍,劉虹宇,曹佩雪,等.HPLC同時測定21種淫羊藿中朝藿定 C和淫羊藿苷的含量[J].藥物分析雜志,2011,31(5):137-140.
[3]張玉萱,徐玲玲.淫羊藿總黃酮的藥理作用研究進展[J].實用臨床醫(yī)藥雜志,2012,16(9):125-128.
[4]蔡曼玲,季暉,李萍,等.5種淫羊藿黃酮類成分對體外培養(yǎng)成骨細胞的影響[J].中國天然藥物,2004,2(4):235-238.
[5]雷永濤,梁妍,郝小燕,等.不同產(chǎn)地淫羊藿中四種活性成分含量的高效液相色譜法測定[J].時珍國醫(yī)國藥,2013,24(6):1404-1405.
[6]國家藥典委員會.中華人民共和國藥典(2010年版)一部[S].北京:中國醫(yī)藥科技出版社,2010:155,306.
[7]郭寶林,肖培根.中藥淫羊藿主要種類評述[J].中國中藥雜志,2003,28(4):303-307.
