高效液相色譜法測定呋塞米片的含量
2014-11-08 08:36:02朱葉青
中國藥業(yè) 2014年13期
朱葉青
(內(nèi)蒙古自治區(qū)呼和浩特食品藥品檢驗所,內(nèi)蒙古 呼和浩特 010020)
呋塞米片是常用的利尿藥,收載于2010年版《中國藥典》,原標(biāo)準(zhǔn)中呋塞米的含量測定采用分光光度法[1]。本試驗中采用高效液相色譜(HPLC)法,結(jié)果證實,該方法準(zhǔn)確、高效、可行。
1 儀器與試藥
LC-2010A型高效液相色譜儀(日本島津公司)。呋塞米對照品(中國藥品生物制品檢定所,批號為100544-200501);四氫呋喃供試品;乙腈為色譜純,其余試劑均為分析純。
2 方法與結(jié)果
2.1 色譜條件與系統(tǒng)適用性試驗
色譜柱:CAPCELL PAK C18MGⅡ柱(250 mm ×4.6 mm,5 μm);流動相:水-四氫呋喃-冰醋酸(60∶40∶0.8);柱溫:30℃;檢測波長:272 nm;進(jìn)樣量:20 μL。理論板數(shù)不低于4 000。在此條件下,色譜圖見圖1。
2.2 溶液制備
取本品20片,精密稱定,研細(xì),精密稱取細(xì)粉適量(約相當(dāng)于呋塞米10 mg),置100 mL容量瓶中,加混合溶劑[冰醋酸22 mL,加乙腈-水(1∶1)至1 000 mL]適量,充分振搖使呋塞米溶解,并稀釋至刻度,搖勻,用0.45 μm的濾膜濾過,取續(xù)濾液作為供試品溶液。精密稱取呋塞米對照品適量,同法制備對照品溶液。
2.3 方法學(xué)考察
陰性干擾試驗:按處方比例制備不含呋塞米的空白片,按供試品溶液制備方法制成陰性對照品溶液,按擬訂色譜條件進(jìn)樣分析。結(jié)果該溶液在呋塞米峰對應(yīng)位置無干擾,見圖1。
線性關(guān)系考察:精密稱取呋塞米對照品10.66 mg,置100 mL容量瓶中,用混合溶劑溶解并稀釋至刻度,搖勻,作為對照品溶液,分別進(jìn)樣 2,10,20,30,40,50 μL,按擬訂色譜條件測定。以峰面積(Y)對進(jìn)樣量(X)進(jìn)行回歸分析,得回歸方程 Y=3.94×106X+8.45×104,r=0.999 9。結(jié) 果 表 明 ,呋 塞 米 進(jìn) 樣 量 在0.212 ~5.33 μg范圍內(nèi)與峰面積呈良好線性關(guān)系。
精密度試驗:取同一呋塞米對照品溶液,重復(fù)進(jìn)樣5次。結(jié)果呋塞米平均峰面積為8 565 608,RSD為0.07%(n=5),表明儀器精密度良好。
穩(wěn)定性試驗:取同一供試品溶液,每間隔1 h進(jìn)樣,共進(jìn)樣6次。結(jié)果平均峰面積為8 566 783,RSD為0.18%(n=6),表明供試品溶液在6 h內(nèi)穩(wěn)定。
重復(fù)性試驗:取同一批樣品,精密稱取5份,照樣品測定項下方法測定。結(jié)果呋塞米的平均含量為97.46%,RSD為0.3%(n=5),表明方法重復(fù)性良好。

圖1 高效液相色譜圖
加樣回收試驗:精密稱取已知含量的樣品9份,精密加入呋塞米對照品適量,按含量測定方法測定,計算回收率。結(jié)果見表1。
2.4 樣品含量測定
取 3 批(批號分別為 1101003,1101007,100701,分別由江蘇亞邦愛普森藥業(yè)有限公司、天津力生制藥股份有限公司、安徽云鵬制藥有限公司生產(chǎn))樣品,按2010年版《中國藥典(二部)》中方法測定其含量,3 批樣品含量分別為 99.2% ,95.7% ,92.3%;而用 HPLC 法測定,其含量分別為 97.4%,92.8%,92.7% 。
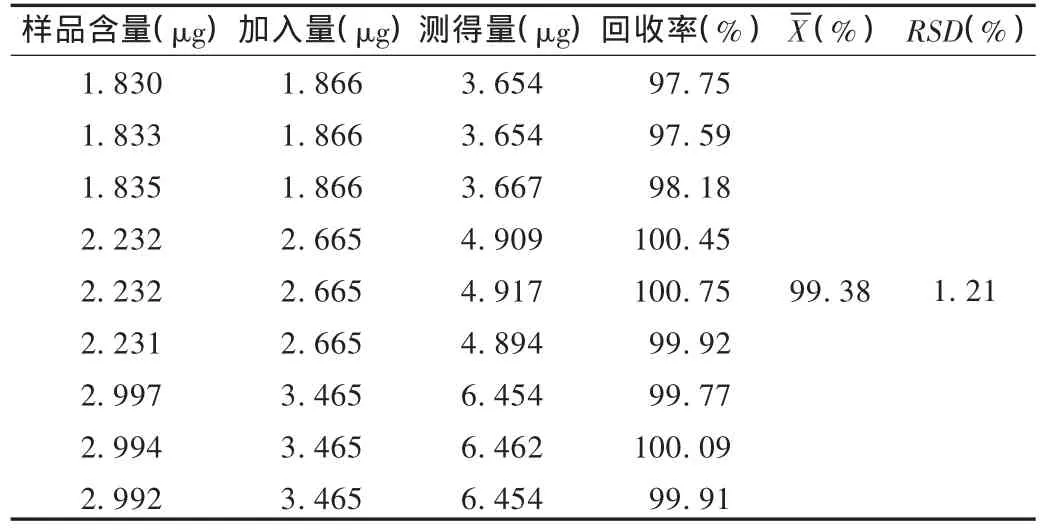
表1 呋塞米加樣回收試驗結(jié)果(n=9)
3 討論
取呋塞米對照品溶液,在200~300 nm波長處進(jìn)行光譜掃描,結(jié)果在272 nm波長處有最大吸收,故選擇272 nm為測定波長。曾采用不同比例的水-四氫呋喃-冰醋酸和甲醇-水-磷酸鹽系統(tǒng)作為流動相進(jìn)行分離[2-3],結(jié)果表明,采用水-四氫呋喃-冰醋酸(60∶40∶0.8)作為流動相時,保留時間適宜,理論板數(shù)較高,分離效果好。
[1]國家藥典委員會.中華人民共和國藥典(二部)[M].北京:中國醫(yī)藥科技出版社,2010:336.
[2]王 峰,陶巧鳳.HPLC法測定呋塞米注射液中的有關(guān)物質(zhì)及含量[J].藥物分析,2012,32(6):1 081-1 084.
[3]陳慶偉,陳華鋒.高效液相色譜法測定呋塞米片的含量[J].海峽藥學(xué),2006,18(2):54.
