重松節(jié)油合成倍半萜烯樹脂的研究
2015-02-23 09:08:51楊韶平陳鍵泉
生物質化學工程 2015年6期
關鍵詞:催化劑
楊韶平, 陳鍵泉, 王 飛, 王 堯
(1.南京林業(yè)大學 化學工程學院,江蘇 南京 210037; 2.梧州學院 化學工程學院,廣西 梧州 543002;3.梧州市嘉盈樹膠有限公司, 廣西 梧州 543001)
·研究報告——生物質材料·
重松節(jié)油合成倍半萜烯樹脂的研究
楊韶平1,2,3, 陳鍵泉3, 王 飛1*, 王 堯3
(1.南京林業(yè)大學 化學工程學院,江蘇 南京 210037; 2.梧州學院 化學工程學院,廣西 梧州 543002;3.梧州市嘉盈樹膠有限公司, 廣西 梧州 543001)
以蒸餾提純后長葉烯和β-石竹烯含量較高的重松節(jié)油為原料、甲苯為溶劑、無水三氯化鋁和引發(fā)劑P為催化劑,經聚合反應生成倍半萜烯樹脂。考察了不同條件對反應的影響,結果表明優(yōu)化的聚合反應條件為:以長葉烯與β-石竹烯總GC含量85%以上的重松節(jié)油為原料,溶劑與原料質量比為0.8∶1,催化劑用量(以原料質量計)4%,反應時間4 h,反應溫度-5~0 ℃。此條件下,樹脂得率為83.7%,軟化點為105 ℃,加納色號為4,長葉烯與β-石竹烯的轉化率達99%以上。
重松節(jié)油;長葉烯;β-石竹烯;蒎烯;萜烯樹脂
近年來,受到石油價格不斷下跌的影響,很多使用蒎烯作為原料生產萜烯樹脂的廠家在成本控制方面未能達到理想效果而被迫停產。雖然這兩年松節(jié)油的產量有下降的趨勢,但重松節(jié)油由于沒有很好的利用途徑,且一直沒有開發(fā)出附加值更高的產品,其保有量多年來不降反增。因此,重松節(jié)油一直被作為低級溶劑和燃料而低價處理,目前其價格還不到蒎烯價格的25%。經研究發(fā)現(xiàn)重松節(jié)油的組成比較復雜,至少由40種以上物質組成,其中含有少量的單萜、醇類、氧化物和低聚物,主要以倍半萜為主[1-3]。通過蒸餾可獲得長葉烯和β-石竹烯含量比較高的重松節(jié)油,由于這些倍半萜類物質含有活潑雙鍵[4-5],與蒎烯具有相似的活性,在一定的反應條件下容易發(fā)生聚合反應生成倍半萜烯樹脂。本研究以長葉烯和β-石竹烯為主要成分的重松節(jié)油為原料合成倍半萜烯樹脂,旨在用于部分替代價格較高的蒎烯合成的萜烯樹脂供下游產品使用。
1 實 驗
1.1 原料及試劑
重松節(jié)油來源于蒸餾完蒎烯的塔釜殘液,廣西梧松林化集團有限公司提供;無水三氯化鋁,引發(fā)劑P,甲苯(符合GB/T 684—1999,分析純),甘油(符合GB/T 13206—2011,分析純),均購買于國藥試劑有限公司。
1.2 原料提純
在實驗室用φ25的玻璃分餾柱對重松節(jié)油進行減壓蒸餾,得提純重松節(jié)油。蒸餾原料:重松節(jié)油;收集餾分:95~100 ℃(2~3 kPa)的餾分。用提純的重松節(jié)油為原料再進行減壓間歇蒸餾,收集長葉烯和β-石竹烯總GC含量分別為55%、65%、75%、85%、95%的餾分,作為聚合反應實驗原料。由于通過分餾很難將長葉烯和β-石竹烯餾分有效分開,長葉烯和β-石竹烯基本上是按3∶1~5∶1的比例共同存在于各餾分中。
1.3 聚合反應
在帶攪拌器、溫度計的500 mL三口燒瓶中,根據(jù)設計的實驗方案加入40~180 g甲苯溶劑,無水三氯化鋁和引發(fā)劑P按質量比4∶1稱取2~7 g研磨粉碎作催化劑,在攪拌的情況下加入到溶劑中,攪拌15~20 min,用實驗室制冷設備進行降溫,稱取100 g蒸餾出來的長葉烯和β-石竹烯總GC含量為55%~95%的重松節(jié)油作為原料,倒進恒壓漏斗中,等溶劑溫度降至-15 ℃時開始滴加。滴加過程中控制好滴料時的溫度,保持溫度在設定的范圍內,攪拌速度為250~300 r/min,滴完后控制好聚合溫度繼續(xù)攪拌反應2~7 h。反應結束后,加入熱水破壞催化劑。
1.4 水解反應
聚合反應結束后進行水解,加入2倍體積量的熱水,破壞催化劑,使三氯化鋁生成易溶于水的氫氧化鋁除去,引發(fā)劑P溶解于水中排去。再用2倍體積量的熱水洗滌聚合液,溫度控制在85~90 ℃,加水后攪拌30 min,靜止分層,分去水層和中層絮狀物,洗滌4次,直到水層呈中性,分析無鋁離子、鐵離子和氯離子為止。
1.5 蒸餾
在常壓下,通過塔釜控制好溶劑回收塔的蒸發(fā)量,直到塔底溫度升至200 ℃,蒸出甲苯和部分未聚合的重松節(jié)油,當基本沒有蒸出物時,控制真空度為2~10 kPa和溫度在230 ℃,繼續(xù)蒸出未聚合的重松節(jié)油,然后升高溫度至250 ℃,并保持真空度為2~10 kPa,開始通入蒸汽活氣蒸餾帶出低聚合度的液體樹脂,最后降溫至200 ℃放出成品固體樹脂。出料前,必須停止抽真空,用氮氣排空至常壓,避免在高溫下由于空氣進入鍋內樹脂氧化變色,與剩余的甲苯或低聚物引起自燃危險。冷卻后得固體樹脂。聚合反應后的產物經水洗后進行蒸餾,收集重松節(jié)油餾分進行色譜分析,計算出未反應的長葉烯、β-石竹烯的含量。
1.6 產物的分析與檢測
1.6.1 氣相色譜分析 福立GC9790氣相色譜儀,OV1701石英毛細管柱(30 m×0.32 mm×0.25 μm),F(xiàn)ID檢測器,檢測器溫度280 ℃,進樣器溫度250 ℃,柱箱溫度為程序升溫至80 ℃,保持5 min,然后繼續(xù)升溫至240 ℃,升溫速度為6 ℃/min,在240 ℃保持15 min[6-7]。
1.6.2 軟化點檢測 依據(jù)GB/T 8146—2003中環(huán)球法進行。將指示溫度計改為內標式,浸沒高度為55 mm,尾長100 mm,刻度范圍50~150 ℃,最小分度為0.2 ℃,加熱介質為甘油。
1.6.3 顏色檢測 將除去外表部分并粉碎好的試樣與甲苯按質量比1∶1溶解后裝入潔凈干燥的比色管中,按照GB/T 1722—1992中鐵鈷比色法進行測定。
1.6.4 樹脂得率計算 固體樹脂得率=固體萜烯樹脂產品質量/原料質量×100%。
2 結果與分析
2.1 溶劑與原料質量比對聚合反應的影響
在催化劑用量(以原料質量計,下同)4%,反應溫度-5~0 ℃,反應時間4 h,長葉烯和β-石竹烯總GC含量85%的重松節(jié)油為原料條件下,改變溶劑與原料質量比,考察其對成品的顏色、樹脂得率和軟化點的影響,結果如表1所示。

表1 溶劑與原料質量比對反應的影響
1)長葉烯和β-石竹烯轉化率,下表同 the conversion rate of longifolene andβ-caryophyllene, the same as those in the following tables
從表1可以看出,隨著溶劑的不斷增多,反應的聚合度也隨之增大,樹脂得率、軟化點和顏色都得到提高,溶劑與原料配比超過0.8∶1以后,陽離子中間體的濃度達到了飽和狀態(tài),溶劑量再增多,其對聚合度的影響不大,樹脂得率、軟化點和顏色的提高都不是很明顯[8]。因此,從經濟角度考慮,溶劑與原料的質量比在0.8∶1~1.0∶1的范圍比較合適。
2.2 催化劑用量對聚合反應的影響
在溶劑與原料質量比為0.8∶1,反應溫度為-5~0 ℃,反應時間為4 h,長葉烯和β-石竹烯總GC含量為85%的重松節(jié)油原料條件下,考察催化劑用量對成品的顏色、樹脂得率和軟化點的影響,結果如表2所示。
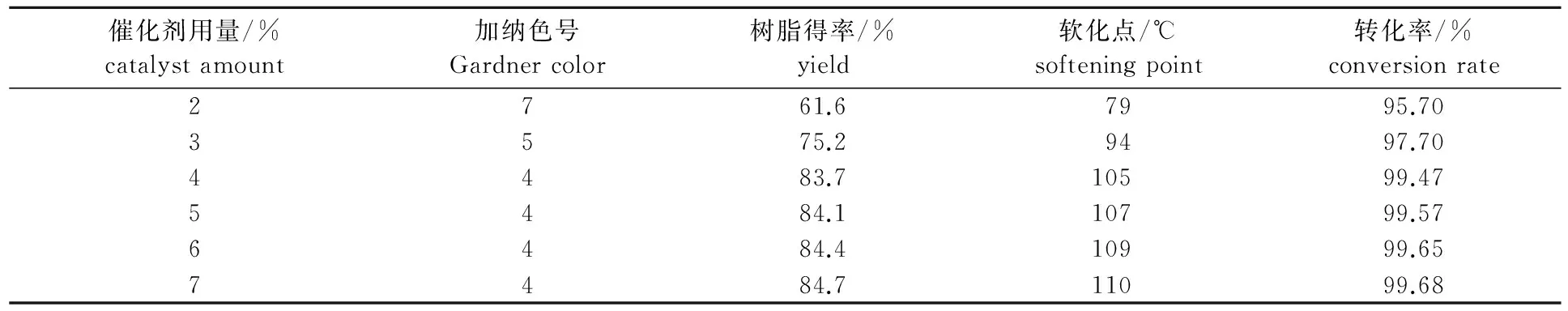
表2 催化劑用量對反應的影響
從表2可以看出,隨著催化劑用量的不斷增多,反應體系中提供給聚合物聚合的H+濃度不斷增大,反應的聚合度也隨之增大,樹脂得率、軟化點和顏色都得到提高[9],在催化劑用量超過4%以后,H+濃度達到了飽和狀態(tài),催化劑用量再增多,其對聚合度的影響都不大,樹脂得率、軟化點和顏色的提高都不是很明顯。因此,從經濟角度考慮,催化劑用量控制在4%比較適宜。
2.3 原料中長葉烯和β-石竹烯含量對聚合反應的影響
在溶劑與原料質量比為0.8∶1,反應溫度為-5~0 ℃,反應時間為4 h,催化劑用量為4%條件下,采用不同長葉烯和β-石竹烯總GC含量的重松節(jié)油原料進行聚合反應,考察其對成品的顏色、樹脂得率和軟化點的影響,結果如表3所示。
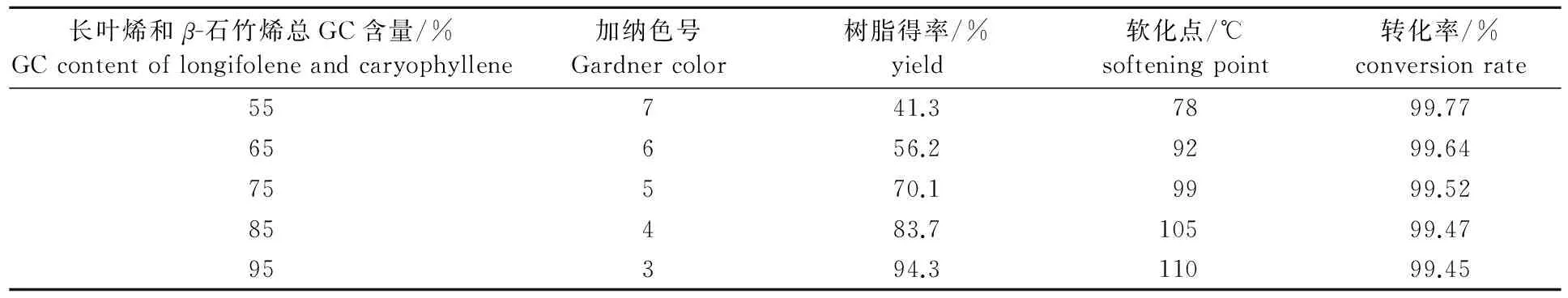
表3 原料中長葉烯和β-石竹烯含量對聚合反應的影響
從表3可以看出,隨著長葉烯和β-石竹烯總GC含量增加,其發(fā)生聚合反應的效果越好。重松節(jié)油中某些萜類物質雙鍵的活潑性差,容易聚合成二聚物等低聚物的液體樹脂[10],從而大大降低成品的軟化點、顏色和固體樹脂產品得率。因此重松節(jié)油中長葉烯和β-石竹烯GC含量越高對反應越有利,由于在重松節(jié)油中蒸餾出高含量的長葉烯和β-石竹烯比較困難,從能耗和產量等經濟因素綜合考慮,聚合原料中長葉烯和β-石竹烯的總GC含量控制在85%以上為宜。
2.4 反應溫度對聚合反應的影響
在溶劑與原料質量比為0.8∶1,催化劑用量為4%,反應時間為4 h,長葉烯和β-石竹烯總GC含量為85%的重松節(jié)油原料條件下,考察反應溫度對成品的顏色、固體樹脂得率和軟化點的影響,結果如表4所示。
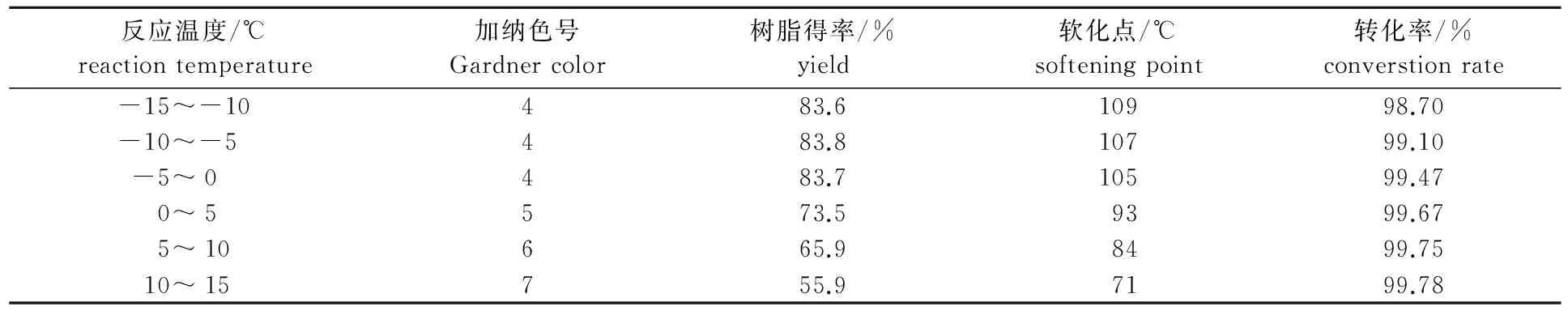
表4 反應溫度對聚合反應的影響
從表4可以看出,隨著反應溫度的不斷升高,原料在H+和溶劑的作用下發(fā)生異構的概率不斷加大,由于聚合的原料具有活潑的雙鍵,容易異構為具有穩(wěn)定雙鍵的其他物質,這些物質多數(shù)反應生成低聚物類液體樹脂等副產物[11],從而大大降低固體樹脂的得率和軟化點。因此,必須控制聚合反應在低溫下進行,但低到0 ℃以下,樹脂得率、軟化點和顏色提高較緩慢,從能耗角度考慮,聚合反應溫度控制在-5~0 ℃比較合適。
2.5 反應時間對聚合反應的影響
在溶劑與原料質量比為0.8∶1,反應溫度為-5~0 ℃,催化劑用量為4%, 長葉烯和β-石竹烯總GC含量為85%的重松節(jié)油原料條件下,考察反應時間對成品的顏色、固體樹脂得率和軟化點的影響,結果如表5所示。
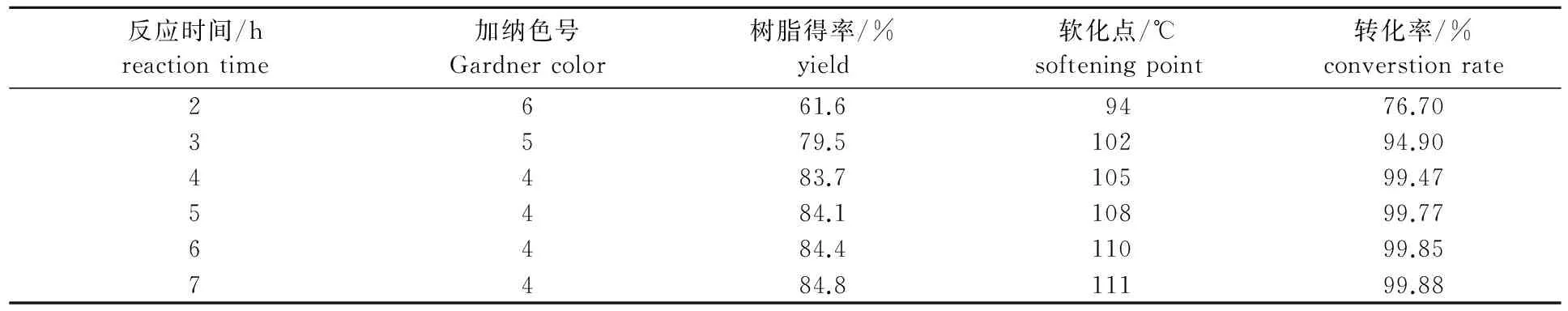
表5 反應時間對聚合反應的影響
從表5可以看出,聚合反應的初期,由于原料濃度高,樹脂濃度低,根據(jù)反應動力學原理,正反應比較快; 4 h以后,反應原料濃度已經降到相當?shù)停系霓D化率已經達到了較高的程度,基本很難反應了。因此,從經濟角度考慮,聚合反應時間為4 h比較合適。
在上述優(yōu)化條件下,固體樹脂得率為83.7%,軟化點為105 ℃,加納色號為4,原料中長葉烯和β-石竹烯轉化率達99%以上,而特級萜烯樹脂(參見LY/T 1453—2010)的加納色號不高于5,軟化點不低于80 ℃,因此,由重松節(jié)油合成的萜烯樹脂的顏色和軟化點主要技術指標符合特級萜烯樹脂的標準。
3 結 論
以蒸餾提純后長葉烯和β-石竹烯含量較高的重松節(jié)油為原料,經聚合反應生成倍半萜烯樹脂,考察了不同條件對反應的影響。結果表明:優(yōu)化的聚合反應條件為:以長葉烯與β-石竹烯總GC含量85%以上的重松節(jié)油為原料,溶劑與原料質量比為0.8∶1,催化劑用量(以原料質量計)為4%,反應時間4 h,反應溫度-5~0 ℃。此條件下,樹脂得率為83.7%,軟化點為105 ℃,加納色號為4,長葉烯與β-石竹烯的轉化率達99%以上。固體樹脂的顏色和軟化點等主要技術指標可達到由蒎烯合成得到的萜烯樹脂的指標要求(LY/T 1453—2010)。通過該方法生產的倍半萜烯樹脂質量指標與蒎烯合成的萜烯樹脂相似,若經過應用性能測試及下游產品應用試驗后有望成新型的樹脂產品獲得應用。
[1]梁志勤,姜紫榮.馬尾松松脂重質松節(jié)油化學組成的研究[J].林產化學與工業(yè),1981,1(2):40-48.
[2]常東亮,哈成勇.松香產品中重質松節(jié)油成分的氣相色譜/質譜法分析[J].分析化學,1999,27(4):423-426.
[3]劉震,程芝.馬尾松松節(jié)油倍半萜成分的分析[J].林產化學與工業(yè),1994,14 (1):21-26.
[4]陳文定,張可欽,王發(fā)松.利用中國重質松節(jié)油合成高軟化點聚倍半萜烯樹脂[J].林產化學與工業(yè),1983,3(4):1-16.
[5]劉莉玫,鄭振德,劉海洋.用雜多酸催化處理的重質松節(jié)油組成分析[J].廣州化學,1995(4):31-35.
[6]徐徐,王琳琳,陳小鵬,等.內標法定量分析α-蒎烯、β-蒎烯、對傘花烴和長葉烯[J].生物質化學與工程,2008,42(1):31-33.
[7]古研,畢良武,趙振東,等.松節(jié)油標準樣品的制備及特征組分研究[J].生物質化學與工程,2006,40(6):1-5.
[8]王婧,趙振東,李冬梅,等.松節(jié)油聚合工藝改進及高品質萜烯樹脂制備研究[J].生物質化學與工程,2012,46(6):7-11.
[9]熊德元.淺色與無色蒎烯樹脂的合成工藝及性能研究[D].南寧:廣西大學博士學位論文,2006.
[10]汪強.改性萜烯樹脂合成工藝及性能研究[D]. 南京:南京林業(yè)大學碩士學位論文,2010.
[11]蘇濤,黃佩芳,肖孝輝,等.用松節(jié)油或重松節(jié)油制備高軟化點萜烯酚醛樹脂[J].林產化學與工業(yè),1998,18 (3):39-48.
歡迎訂閱2016年《中國造紙》
《中國造紙》(月刊)為專業(yè)技術性刊物,國內外公開發(fā)行,由中國造紙學會和中國制漿造紙研究院主辦,主要報道我國造紙工業(yè)在原材料、制漿、造紙、廢液綜合利用及污染防治、機械設備、分析檢驗、工藝和質量控制自動化以及制漿造紙專業(yè)基礎理論等方面的新成就和重要科技成果。除及時報道各研究機構、高等院校在科研理論方面取得的突破成果外,還注重報道各制漿造紙廠引進或自行研究探索的新工藝、新技術。該刊將理論與實踐有機結合,更好地滿足了科研工作者以及制漿造紙工廠技術人員的需要。該刊是我國造紙界權威性技術期刊,連續(xù)入選“中文核心期刊”、“中國科技論文統(tǒng)計源期刊”、“中國科學引文數(shù)據(jù)庫來源期刊”、“中國科學文獻評價數(shù)據(jù)來源期刊”,并已被Scopus、CA等國外著名的期刊索引收錄。入選“中國科協(xié)精品科技期刊工程第四期項目”。
刊號CN 11-1967/TS, ISSN 0254-508X,每月25日出版,大16開,國內單價:10元/冊,全年120元;國外及港澳臺地區(qū):20美元/冊,全年240美元。國內總發(fā)行:北京市報刊發(fā)行局,郵發(fā)代號:2-194;國外總發(fā)行:中國出版對外貿易總公司,發(fā)行代碼:DK11070。全國各地郵局均可訂閱,如錯過郵局訂閱,可直接與中國造紙雜志社發(fā)行部聯(lián)系補訂。電話:010-64778173(發(fā)行部),010-64778158~61(編輯部);傳真:010-64778174;E-mail: cpp2108@vip.163.com;通信地址:100102北京朝陽區(qū)望京啟陽路4號中輕大廈6層中國造紙雜志社。
Synthesis of Sesquiterpene Resin from Heavy Turpentine
YANG Shao-ping1,2,3, CHEN Jian-quan3, WANG Fei1, WANG Yao3
(1.School of Chemical Engineering,Nanjing Forestry University, Nanjing 210037, China; 2.School of Chemical Engineering,Wuzhou University, Wuzhou 543002, China; 3.Wuzhou Cayin Gum Ltd., Wuzhou 543001, China)
Sesquiterpene resin was prepared from heavy turpentine with high content of longifolene andβ-caryophyllene after being purified by distillation. In the reaction, the xylene was solvent, and anhydrous aluminium chloride and initiator P were used as catalyst. The optimum reaction conditions were investigated. The results showed that the yield, softening point and Gardner color of the product were 83.7%, 105 ℃ and 4, respectively. The conversion rate of longifolene andβ-caryophyllene was over 99%, when the GC content of longifolene andβ-caryophyllene in the heavy turpentine was over 85%, the weight ratio of solvent to heavy turpentine was 0.8∶1, the dosage of catalyst was 4%, the reaction time was 4 h and reaction temperature was -5-0 ℃, respectively.
heavy turpentine; longifolene;β-caryophyllene; pinene; terpene resin
10.3969/j.issn.1673-5854.2015.06.004
2015- 07- 09
廣西高校科學技術研究項目(ZD204123,KY2015WZ004);江蘇省博士后基金(1401010A);梧州學院校級科研項目(2011B001)
楊韶平(1976—),男,江西撫州人,副教授,博士,從事松節(jié)油的研究;E-mail:ysp1120@163.com
*通訊作者:王 飛(1962—),教授,博士生導師,主要從事林產化學品研究;E-mail:hgwf@njfu.edu.cn。
TQ35
A
1673-5854(2015)06- 0017- 05
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
