DNA甲基化檢測技術進展及經驗總結
2015-03-03 07:06:54李光潤綜述審校
醫學綜述 2015年2期
關鍵詞:檢測
李 懿,李光潤(綜述),陳 宏(審校)
(成都軍區昆明總醫院腫瘤科,昆明 650032 )
?
DNA甲基化檢測技術進展及經驗總結
李懿,李光潤(綜述),陳宏※(審校)
(成都軍區昆明總醫院腫瘤科,昆明 650032 )
摘要:DNA甲基化作為一種表觀遺傳學元件,參與正常細胞的發育分化、基因組印記、X染色體失活和染色質重構等生命過程,在腫瘤演進中亦發揮重要作用。為滿足不斷深入的研究需求,DNA甲基化檢測技術得到了較快的發展,檢測的靈敏度和特異度不斷提高,并逐步從個別位點向高通量檢測發展。該文綜述了目前常用的DNA甲基化分析方法,并簡要總結了其在應用中的關鍵點和經驗。
關鍵詞:表觀遺傳學;DNA甲基化;檢測
DNA甲基化是表觀遺傳學修飾的主要形式,甲基化模式的異常促進細胞惡性轉化,參與腫瘤演進[1-2]。許多基因具有腫瘤特異性的甲基化表型[3],一方面基因組廣泛的低甲基化引起原癌基因活化,基因組不穩定性增加[4];另一方面啟動子區的高甲基化,引起抑癌基因及錯配修復基因表達沉默,導致腫瘤的發生[5]。研究表明,基因組的低甲基化隨著細胞惡性度的增加而愈加明顯,是病情診斷的重要指標[6];此外,CpG島局部的高甲基化發生于細胞惡變之前,能在早期預測腫瘤的發生[7],其也能有效地預測腫瘤的復發和轉移[8],在腫瘤的鑒別診斷中亦發揮重要作用[9]。因此,檢測腫瘤發生中相關基因的甲基化模式具有重要意義,現就近年來中外文獻報道的甲基化檢測方法進行簡要的分析總結。
1酶切-Southern雜交技術
酶切-Southern雜交是早期用于甲基化研究的技術,其依賴于同裂酶(HpaⅡ和MspⅠ)識別位點中甲基胞嘧啶的敏感度不同,與基因組DNA共同消化后,與32P標記的檢測基因片段的探針行Southern雜交,根據片段長度分辨位點的甲基化情況。該方法簡單且實驗結果易解釋,可進行基因CpG島總體甲基化模式的定量研究,但樣品需要量大且僅提供位于甲基化敏感限制性內切酶識別序列中的CpG甲基化信息,易出現由不完全酶切引起的假陽性。
2亞硫酸氫鈉修飾后測序法
亞硫酸氫鈉修飾后測序法是一種對DNA進行亞硫酸氫鈉處理、聚合酶鏈反應(polymerase chain reaction,PCR)擴增與DNA測序相結合的方法,能夠提供測定區域的序列信息,準確定位甲基化胞嘧啶位點;重亞硫酸鹽修飾后,甲基化胞嘧啶保持不變,但非甲基化胞嘧啶轉變為尿嘧啶,PCR擴增后為胸腺嘧啶,其將甲基化狀態的差異轉化成堿基的差異,從而對胞嘧啶的甲基化狀態進行分析;但在亞硫酸鈉處理的酸性環境下,單鏈特異性PCR模板穩定性下降,容易降解;并且模板鏈CG二核苷酸水平高易形成復雜的二級結構,常出現非特異性條帶,結合“巢式PCR法”能顯著提高擴增的特異度[10]。在運用亞硫酸氫鈉修飾后測序法中要注意如下5個方面。①DNA的質量控制:DNA模板純度要求高,含量約2 μg,否則在亞硫酸鹽處理時會因DNA模板太多致使DNA處理不完全進而導致PCR擴增失敗,如果DNA模板太少會導致假陰性。②修飾時的溫度控制:溫度過低或過高都會導致DNA模板處理失敗,所用試劑一定要現用現配,嚴格按試劑盒說明書進行,保證試劑的pH值為8.0,否則會降低修飾效率,修飾后的模板應保存在-20 ℃,不應超過2個月。③引物的合理設計:在亞硫酸氫鈉修飾后測序法中引物設計是關鍵,引物設計時應遵循如下原則:a.主要針對編碼鏈設計引物,以修飾后的DNA序列作為模板;b.選擇CpG島作為PCR擴增的靶區域;c.引物根據不含CpG位點的序列來設計,但擴增區域中應包括盡可能多的CpG位點。④退火溫度的控制:在PCR反應中,CG含量高的退火溫度值高,非甲基化引物中正義引物不含C,反義引物不含G。⑤反應增強劑:當CG含量高于70%或模板中有二級結構時可考慮添加增強劑,從而減低G+C富集區的熔點及解鏈溫度、提高擴增效率,并增加Taq DNA連接酶的穩定性,防止其因溫度升高而變性。但一定要注意它們的濃度范圍(二甲基亞砜 1%~10%,甘油5%~20%),超過一定濃度對酶有抑制作用,反而使擴增效率降低。
3甲基化特異性PCR
MS-PCR以亞硫酸鈉修飾的甲基化依賴的單核苷酸多態性為基礎,設計能區分基因組甲基化狀態的引物,識別亞硫酸鈉修飾的甲基化和非甲基化等位基因的序列差異,同時也識別沒有修飾DNA和不完全修飾DNA的序列差異,通常選擇胞嘧啶含量高的序列設計引物,引物的3′端鄰近檢測的CpG二核苷酸,引物覆蓋序列中CpG島所占比例越高,甲基化DNA檢出率越高。該方法簡單、省時、敏感,還可用于石蠟包埋樣品甲基化分析,能檢測出樣品中0.1%的甲基化DNA含量[11]。為提高甲基化的檢出率,對甲基化特異性PCR(methylation-specific PCR, MS-PCR)進行改進后設計出半巢式PCR和巢式PCR。其中,半巢式PCR是指用甲基化引物MS、M2A和非甲基化引物US、U2A分別進行甲基化和非甲基化的特異性擴增;然后在MS和M2A之間增加一條引物M1A,再以MS、M1A進行第二輪甲基化特異性擴增,同樣的用US、U1A作第二輪的非甲基化擴增。而巢式PCR是指用非特異性的外側引物對處理后的樣品進行擴增,然后再用特異性的內側甲基化引物和非甲基化引物做第二輪擴增。用MS-PCR擴增后,可能會出現如下3種可能的結果:①產物電泳為陰性(無目的基因帶出現);②產物電泳出現多條非特異帶(包括目的基因);③產物電泳為陽性(只有目的基因帶)。出現①和②結果時,在保證反應體系和引物沒有問題的情況下可通過調整MS-PCR的反應溫度而得到目的基因帶。按圖1所示的方法,根據電泳結果調整退火溫度[12]。
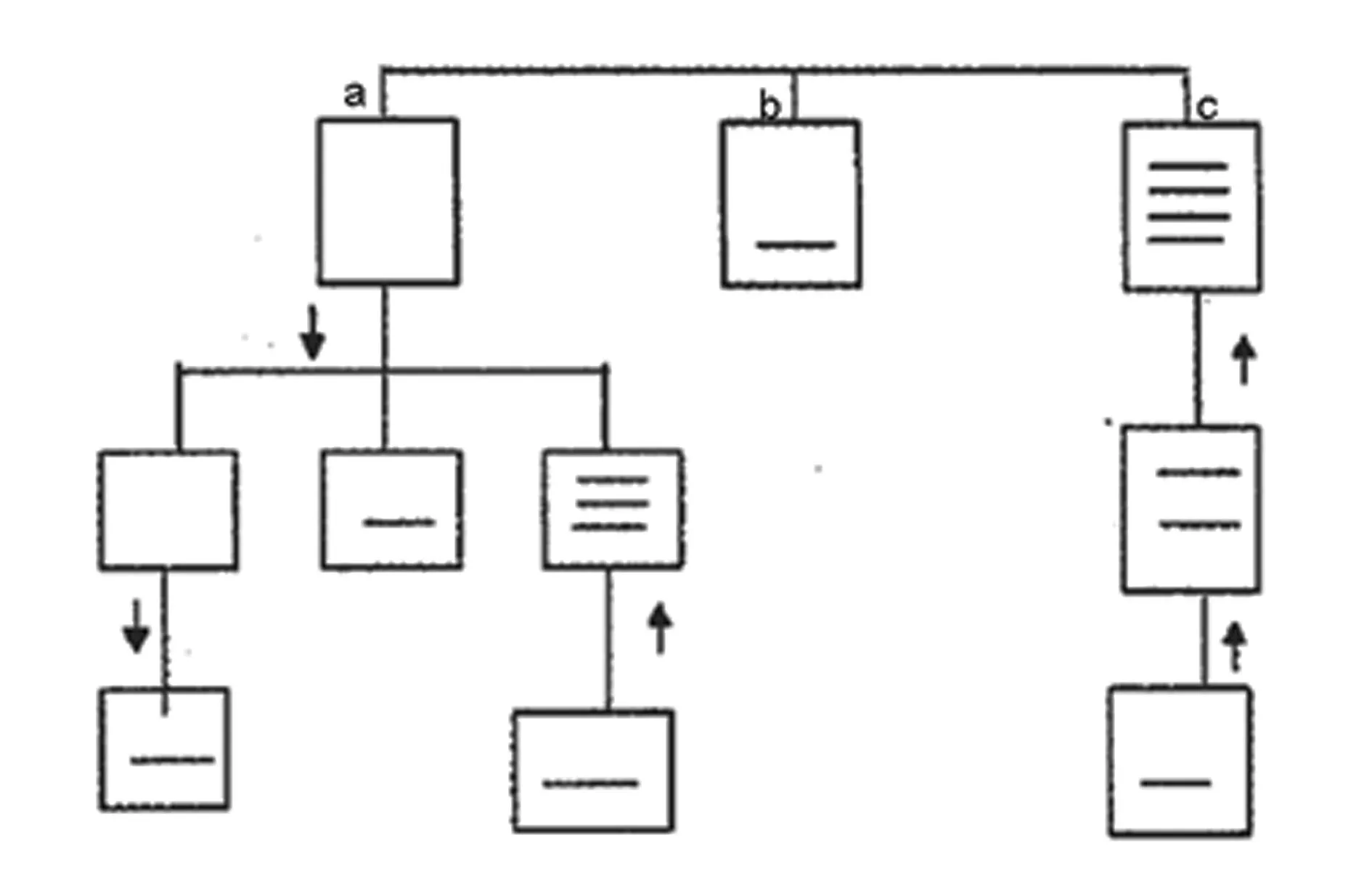
a:產物電泳為陰性(無目的基因帶出現);b:擴增出特異的目的條帶;c:產物電泳出現多條非特異帶(包括目的基因);當出現a和c的情況時,按圖中所示調整退火溫度;↑表示升高退火溫度;↓表示降低退火溫度圖1 甲基化特異性聚合酶鏈反應電泳結果
4結合亞硫酸鹽處理的限制酶解分析法
該技術通過限制性酶切反應揭示亞硫酸鈉處理、PCR擴增后引入的甲基化依賴的單核苷酸多態性。Xiong和Laird[13]闡述了結合亞硫酸鹽處理的限制酶解分析法的基本步驟,用限制性內切酶BstUI識別2個CG二核苷酸均發生甲基化的序列(5mCG5mCG),用限制性內切酶TaqI僅識別1個CG二核苷酸的甲基化,進行限制性切割后,能準確定量樣本的甲基化比例。該方法操作簡單,能準確定量,可分析微切割的石蠟包埋樣品,避免了Southern雜交定量時由甲基敏感性酶產生的假陽性;但其局限性在于只能獲得特殊酶切位點的甲基化情況,不能除外假陰性檢測的可能性。用亞硫酸氫鈉聯合限制性內切酶分析法進行甲基化檢測時應注意:①引物的設計與MS-PCR不同,亞硫酸氫鈉聯合限制性內切酶分析法的引物不能含有CpG雙核苷酸序列;而MS-PCR要有分別針對甲基化和非甲基化的引物,引物中保證有3個以上的CpG雙核苷酸序列。②亞硫酸鈉處理的質量要求高,可通過設立限制性內切酶Hsp9211酶切內對照確保亞硫酸鈉修飾完全。③所有的酶切反應在適宜溫度下至少進行4 h[14]。
5差異甲基化位點掃描
該方法利用低熔點瓊脂糖對基因組DNA的保護作用,使DNA免受物理性損傷,避免DNA分散,使得從少量的細胞(10個細胞/反應)中既可獲取大量DNA進行甲基化檢測,同時在瓊脂糖中進行限制性酶切后,用重復序列引物或隨機引物進行第一輪PCR擴增,利用甲基化和非甲基化模板擴增產物的片段差異,進行第二輪任意引物的擴增,根據變性電泳后擴增條帶的亮度反映模板的甲基化程度[15]。該方法對樣品的需要量非常少,能有效地避免大量樣品間由于細胞異質性引起的差異,結果可信度高,其使單細胞甲基化分析成為可能。
6甲基化熒光檢測法
該方法是在MSP技術的基礎上,結合熒光實時PCR的甲基化定量分析方法。經亞硫酸鈉處理后,需要用雙標記的熒光探針進行擴增,當探針與靶序列完全匹配時,可被具有核酸外切酶活性的Taq DNA多聚酶切割,釋放5′端報告基因,遠離3′端熒光淬滅基團的屏蔽,使得5′端熒光染料受激發所發射的熒光信號可被探頭檢測,熒光信號的大小與擴增產物的量成比例[16]。其中,鎖式探針作為近來常用的一種探針形式,其原理是在完全匹配的前提下,線性鎖式探針被有效地連接成環形,并可以在后續的PCR反應中進行擴增;而沒有配對的序列時,探針只能以線性的形式存在,在核酸外切酶作用下,線性探針被消化水解而不能進行PCR擴增[17]。該方法避免了上述檢測技術中PCR擴增后進行凝膠電泳及限制性酶切的常規操作,在一定程度上提高了檢測的敏感度及特異度,常用于臨床標本的分析。但由于檢測探針的價格較高,且需要依賴昂貴的定量PCR儀,所以檢測的成本較高。
7甲基化特異性寡核苷酸微陣列
依據基因組范圍內分析基因突變和單核苷酸多肽性的微陣列雜交技術原理,發展了甲基化特異性寡核苷酸微陣列,利用模板中甲基化狀態的差異經PCR反應后擴增出不同的核苷酸序列,與微陣列上的能區分特異胞嘧啶位點甲基化狀態的寡核苷酸探針雜交,高通量同步定量分析全基因組的甲基化模式[18]。Gitan等[19]的研究發現,部分甲基化特異性核寡苷酸微陣列檢測為0甲基化的樣品能被MSP的甲基化引物擴增出了一條微弱的條帶,提示雜交背景的信號影響了其對CpG島低甲基化水平的檢測,可通過調整雜交條件和洗片條件來減少假陽性結果。此方法的關鍵是寡核苷酸探針,設計時應注意:①針對2個或多個CpG位點的探針,能顯著減少交叉反應,但應避免連續超過4個胸腺嘧啶殘基或鳥嘌呤殘基;②重疊2個以上的CpG位點,則探針應包括每個位點甲基化和非甲基化胞嘧啶的所有組合;③實驗中應設立明確的對照系統測試寡核苷酸探針的有效性。該方法能形成癌癥遺傳圖譜,具有良好的應用前景[20]。
8變性高效液相層析
將亞硫酸鈉修飾后的DNA分解為單核苷酸后,依據5種核苷酸在極性溶液中的溶解度不同,其在色譜柱中的保留時間存在差異,溶解度高的隨極性溶液先過濾出,而溶解度低的則在柱中的停留時間較長,各核苷酸的出柱順序是:dCp、5′mdCp、Tp、dGp和dAp,從而了解樣品全基因組水平的甲基化程度,但該方法的局限性在于不能了解特定基因的甲基化的位置和狀態,并且對樣品DNA的純度要求高,不能有RNA的污染[21]。
9甲基化DNA免疫共沉淀
利用5-甲基胞嘧啶特異性抗體或含有甲基結合結構域的蛋白,通過免疫沉淀作用于特異性的富集基因組中的甲基化或非甲基化片段。其基本程序是:將基因組DNA打斷為400~500 bp的片段,經加熱變性后將樣品分為2份,其中1份作為Total input DNA 樣品,而另1份加入甲基化DNA特異性抗體,使用親和層析分離上述樣品中的甲基化DNA片段抗體復合物,而洗脫其中的非甲基化片段,純化得到的甲基化DNA可以結合熒光定量PCR技術或芯片技術檢測基因的甲基化[22]。該方法特異度高,精確性好,避免了探針合成,但是對低CpG密度區域的甲基化分析存在問題。
10結語
甲基化檢測技術的不斷進步,為揭示疾病中基因表達沉默的表觀遺傳機制提供了有效的手段,從而能夠高通量地分析多個基因多個CpG位點的甲基化模式,并從總體動態的角度了解疾病相關的甲基化信息。通過介紹近幾年來DNA甲基化檢測的新方法,總結了各方法在應用中的經驗和注意事項希望能對DNA甲基化的研究者有所幫助。同時也期待著甲基化技術的不斷進步,檢測的敏感度和特異度不斷提高,以滿足臨床檢測的要求,使其臨床的應用范圍更加廣泛,能更好地指導臨床的個體化診療。
參考文獻
[1]Sandoval J,Esteller M.Cancer epigenomics: beyond genomics[J].Curr Opin Genet Dev, 2012,22(1):50-55.
[2]Kanwal R,Gupta S.Epigenetic modifications in cancer[J].Clin Genet,2012,81(4): 303-311.
[3]Ma X,Wang YW,Zhang MQ,etal.DNA methylation data analysis and its application to cancer research[J].Epigenomics,2013,5(3):301-316.
[4]Akhavan-Niaki H,Samadani AA.DNA methylation and cancer development:molecular mechanism[J].Cell Biochem Biophys,2013,67(2):501-513.
[5]Fukushige S,Horii A.DNA methylation in cancer: a gene silencing mechanism and the clinical potential of its biomarkers[J].Tohoku J Exp Med,2013,229(3):173-185.
[6]Heichman KA,Warren JD.DNA methylation biomarkers and their utility for solid cancer diagnostics[J].Clin Chem Lab Med,2012,50(10):1707-1721.
[7]Markopoulou S,Nikolaidis G,Liloglou T.DNA methylation biomarkers in biological fluids for early detection of respiratory tract cancer[J].Clin Chem Lab Med,2012,50(10):1723-1731.
[8]Nai HS,Lau QC.Advent of the cancer methylome[J].Comb Chem High Throughput Screen,2012,15(3):216-220.
[9]Szyf M.DNA methylation signatures for breast cancer classification and prognosis[J].Genome Med,2012,4(3):26.
[10]Frommer M,McDonald LE,Millar DS,etal.A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands[J].Proc Natl Acad Sci U S A,1992,89(5):1827-1831.
[11]Licchesi JD,Herman JG.Methylation-specific PCR[J].Methods Mol Biol,2009,507:305-323.
[12]Liu ZJ,Maekawa M.Polymerase chain reaction-based methods of DNA methylation analysis[J].Anal Biochem,2003,317(2):259-265.
[13]Xiong Z,Laird PW.COBRA:a sensitive and quantitative DNA methylation assay[J].Nucleic Acids Res,1997,25(12):2532-2534.
[14]Boyko A,Kovalchuk I.Analysis of locus-specific changes in methylation patterns using a COBRA (combined bisulfite restriction analysis) assay[J].Methods Mol Biol,2010,631:23-31.
[15]Hamada T,Murasawa S,Asahara T.Simple screening method for differentially methylated regions of the genome using a small number of cells[J].Biochem Biophys Res Commun,2007,353(2):275-279.
[16]Eads CA,Danenberg KD,Kawakami K,etal.MethyLight:a high-throughput assay to measure DNA methylation[J].Nucleic Acids Res,2000,28(8):E32.
[17]Deng J,Shoemaker R,Xie B,etal.Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming[J].Nat Biotechnol,2009,27(4):353-360.
[18]Lister R,O′Malley RC,Tonti-Filippini J,etal.Highly integrated single-base resolution maps of the epigenome in Arabidopsis[J].Cell,2008,133(3):523-536.
[19]Gitan RS,Shi H,Chen CM,etal.Methylation-specific oligonucleotide microarray:a new potential for high-throughput methylation analysis[J].Genome Res,2002,12(1):158-164.
[20]Mund C,Beier V,Bewerunge P,etal.Array-based analysis of genomic DNA methylation patterns of the tumour suppressor gene p16INK4A promoter in colon carcinoma cell lines [J].Nucleic Acids Res,2005,33(8):e73.
[21]Ramsahoye BH.Measurement of genome wide DNA methylation by reversed-phase high-performance liquid chromatography[J].Methods,2002,27(2):156-161.
[22]Nair SS,Coolen MW,Stirzaker C,etal.Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias[J].Epigenetics,2011,6(1):34-44.
Progresses and Experiences of DNA Methylation Detection TechniquesLIYi,LIGuang-run,CHENHong.(DepartmentofOncology,KunmingGeneralHospitalofChengduMilitaryCommand,Kunming650032,China)
Abstract:DNA methylation,as an epigenetic component,is involved in the processes of cell development,differentiation,genomic imprinting,X chromosome inactivation and chromatin remodeling,and plays an important role in carcinogenesis as well.The DNA methylation analysis technology has been developing rapidly due to the needs of deeper research:analyses that previously were restricted to specific loci can now be performed on a high throughput,meanwhile the sensitivity and specificity have made great of improvements as well.Here is to make a review of the methods to detect DNA methylation and summarize the key points and experiences in application.
Key words:Epigenetic; DNA methylation; Detection
收稿日期:2014-02-27修回日期:2014-06-23編輯:鄭雪
基金項目:國家自然科學基金(81201756);云南省自然科學基金(2012FD091)
doi:10.3969/j.issn.1006-2084.2015.02.005
中圖分類號:Q3-3; Q751
文獻標識碼:A
文章編號:1006-2084(2015)02-0204-04
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
