誤診為腦梗死的MELAS 1例報告
2015-03-11 03:34:08呂曉民,孫立超,顧金花等
中國實驗診斷學 2015年12期
誤診為腦梗死的MELAS 1例報告
呂曉民,孫立超,顧金花,邱吉慶*
(吉林大學第一醫院,吉林 長春130021)
線粒體腦肌病(mitochondrial encephalomyopathy,ME)為中樞神經系統少見代謝性疾病,是由于線粒體結構與功能障礙、呼吸鏈受損所致的多系統疾病,以骨骼肌及中樞神經系統受累為主。線粒體腦肌病伴乳酸中毒和中風發作 (mitochondrial encephalomyopathy,lactic acidosis,and strokelike episodes ,MELAS)為ME的常見類型,其臨床表現多樣,臨床誤診率高。現將我院診治的1例MELAS報道如下,并復習其相關文獻。
1臨床資料
患者,女,31歲。緣于16個月前無明顯誘因出現抽搐,表現為:四肢強直-陣攣,頭眼向右側偏斜,伴口吐白沫,無舌咬傷及尿失禁,持續約1 min緩解,發作后意識不清,就診吉大一院。頭部MRI示:右側頂枕葉大片狀稍長T1長T2異常信號。診斷為:腦梗死、癲癇。給予改善循環,抗癲癇及對癥治療。經治療后患者神清,自訴發作時視物模糊、惡心。出院后給予卡馬西平100 mg,3/日口服。
9個月前患者再次出現上述發作,再次就診,經左肱二頭肌活檢示:肌纖維直徑70 μm左右,HE染色部分肌纖維肌膜下嗜堿,經GT染色證實為不典型RRF,NADH及CDX染色部分肌纖維肌膜下濃染。診斷為線粒體腦肌病。給予抗癲癇及對癥治療。治療后無癲癇發作。
7個月前患者出現右側肢體不自主摸索樣動作,頭向一側偏斜,以向右側偏斜次數為多,每次持續約30 s,每天發作數十次,發作時患者意識清醒,伴惡心,偶有嘔吐,非噴射性,嘔吐物為胃內容物。再次就診于我院。既往糖尿病1年,最高空腹血糖8.0 mmol/L,未給予系統治療。足月順產,否認顱內感染史、頭部外傷史。否認肝炎、結核等傳染病病史;否認高血壓病、冠心病;否認藥物過敏史。否認手術史。追問其家族史無特殊記載(詳細追問其母系,未見特殊)。查體:發育尚可,無智力低下。右利手,記憶力差,右顳側偏盲,雙側視神經萎縮,視力減弱,R 0.1,L 0.06。查體未見明顯神經系統陽性體征。運動前血乳酸1.5 mmol/L,運動后10 min血乳酸2.1 mmol/L。乳酸脫氫酶463 U/L,肌酸激酶61 U/L。磁共振頭部平掃:左側枕葉可見斑片狀異常信號,T1WI呈稍低信號,T2WI呈稍高信號,Flair呈稍高信號,DWI均為高信號(圖1)。腦電圖:左側枕區無優勢節律。左側枕、后顳區棘波、棘慢波發放,多可波及至同側頂區。監測到數十次左側枕區起源的臨床發作及4次左側枕區起源的單純部分性發作。臨床診斷為:線粒體腦肌病(MELAS綜合征)、癥狀性癲癇(單純部分發作繼發全面強直-陣攣)。給予高蛋白、高碳水化合物、低脂飲食、ATP、輔酶Q10、維生素B1、甲鈷胺等改善能量代謝。同時給予抗癲癇藥物得理多200 mg,3/日口服。經治療后患者癥狀明顯緩解后出院。
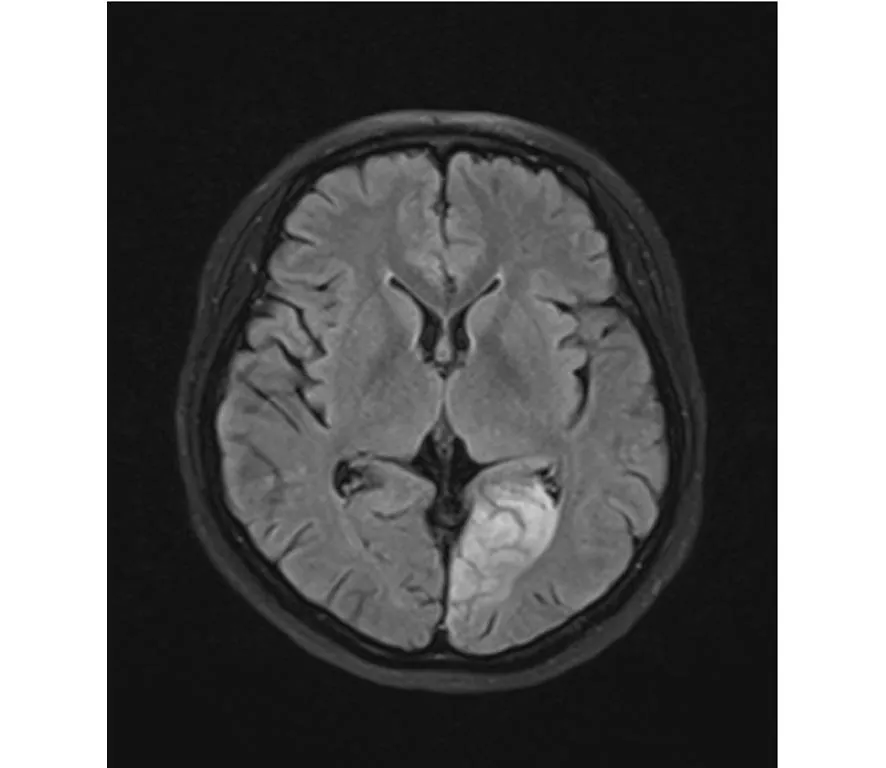
圖1 磁共振頭部平掃:左側頂枕葉Flair呈稍高信號
2討論
線粒體為廣泛存在于真核細胞中的細胞器。主要功能是通過氧化磷酸化作用合成ATP,為細胞各種生理活動提供能量。線粒體肌病為氧化磷酸化脫偶聯引起,是能量代謝障礙所致的骨骼肌極度不能耐受疲勞,病變以侵犯骨骼肌為主。如病變同時侵犯中樞神經系統則為線粒體腦肌疾病。線粒體腦肌病主要分為以下11種類型:(1)MELAS綜合征;(2)MERRF綜合征;(3)KSS綜合征;(4)CPEO綜合征;(5)Leigh病;(6)Alpers病;(7)Menke病;(8)LHON綜合征;(9)NARP綜合征;(10)Wolfram綜合征;(11)MNGIE綜合征。
線粒體腦肌病以MELAS綜合征最為常見,是因線粒體DNA突變所致的逐漸進展的神經變性疾病,呈母系遺傳。可累及多個系統及器官,如神經系統、骨骼肌、內分泌系統、胃腸道、皮膚等[1]。MELAS綜合征于1975年由Shapira首次報道[2]。1984年Pavlakis論述MELAS綜合征的診斷標準[3]。
MELAS綜合征暫無特殊療法,主要集中于系統治療與局部治療兩方面。系統治療為改善能量代謝,如ATP、輔酶Q等。局部治療為對癥治療,如合并癲癇的患者給予抗癲癇藥物,合并偏頭痛的患者給予止痛藥物等。
本例患者以癲癇起病,隨即出現意識障礙,頭部MRI示:右側頂枕葉大片狀稍長T1長T2異常信號。誤診為:腦梗死。經治療后意識恢復。7月后再次出現癲癇發作,經肌肉活檢,發現不典型RRF,NADH及CDX染色部分肌纖維肌膜下濃染。診斷為:線粒體腦肌病。9個月后出現起源于枕葉的癲癇發作,同時行乳酸耐量試驗呈陽性。綜合三次住院的臨床表現及輔助檢查最終診斷為:線粒體腦肌病(MELAS綜合征)、癥狀性癲癇。
綜上,MELAS綜合征臨床表現復雜,易于誤診誤治,對于符合Michio診斷標準的患者應及早行肌肉活檢,或mtDNA檢查,以助早期診斷及治療[4]。
參考文獻:
[1]Kaufman KR,Zuber N,Rueda-Lara MA,et al.MELAS with recurrent complex partial seizures,nonconvulsive status epileptic psychosis,and behavioral disturbances:Case analysis with literature review[J].Epilepsy & Behavior,2010,18(4):494.
[2]Shapira Y,cederbaum SD,cancilla Pa,et al.Familial poliodystrophy,mitochondrial myopathy,and lactate acidemia[J].Neurol,1975,25(7):614.
[3]Pavlakis SG,Phillips PC,Dimauro S,et al.Mitochondrial myopathy,encephalopathy,lactic acidosis,and strokelike episodes:a distinctive clinical syndrome[J].Ann Neurol,1984,16(4):481.
[4]Michio Hirano,Enzo Ricci,M Richard Koenigsberger.MELAS:An original case and clinical criteria for diagnosis[J].Neuromuscular Disorders,1992,2(2):125.
(收稿日期:2014-09-03)
文章編號:1007-4287(2015)12-2139-02
*通訊作者
基金項目:吉林省科技發展計劃項目(編號20110469)
猜你喜歡
中國民間療法(2021年5期)2021-06-09 09:21:04
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
中國生殖健康(2019年3期)2019-02-01 06:12:26
飲食科學(2017年5期)2017-05-20 17:11:53
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
西南軍醫(2015年4期)2015-01-23 01:19:30
中國中醫藥現代遠程教育(2014年20期)2014-03-01 04:31:21
