AMI方法對HIV病毒分類
2015-03-27 12:26:52劉旻昊
山東青年 2015年1期
劉旻昊
摘要:在生物的基因序列中,蘊含了其所有的特點和規律,是大自然隱藏在生物千變萬化形態下的“密碼”。在本文中我們選取21種HIV病毒基因數據,應用基于非序列比對的平均互信息方法提取出它們的序列特征,結合相關系數和離差平方和方法(Ward法)對其進行分類。此種分類方法有別于傳統的序列對比方式,運算簡單,速度快捷且得到了合理的分類結果。
關鍵詞:平均互信息;基因組;離差平方和方法
引言
在醫學領域,從DNA分子水平來研究疾病的起因發展與分類,解讀病毒基因的“密碼”,正日益引起分子生物學者、數學、計算機以及信息網絡科學研究人員的重視。如何分析這些DNA序列數據,提取出能夠量化的“信息”來描述它們之間的聯系,是當前研究的熱門問題。HIV病毒在進化過程中形成了三種亞型分類,目前對這種進化分類常用的方法有最大簡約法、距離矩陣法和最大似然法等。相應的也有一系列軟件,如:PHYLIP、PAUP和MEGA等。通常在應用這些方法之前,都要對序列進行比對(sequence alignment),常用的軟件有CLUSTRALW等。
本文提取不同DNA序列的平均互信息(Average Mutual Information,AMI)[2]作為特征參數,構造AMI向量,通過AMI向量的相關系數定義不同DNA序列之間的距離,利用離差平方和法對距離矩陣進行聚類分析,從而得到他們的進化關系。此種方法是非序列比對方法,計算簡單且速度較快,對大量數據的處理非常方便,在醫學領域中有著廣泛的應用。
1.理論與方法
1.1平均互信息(AMI)
DNA序列是4種核苷酸A、C、G、T的集合,如果x代表在基因序列上某一位置的核苷酸,則y為在x下游方向間隔k個位置的核苷酸。n\-k(x,y)表示核苷酸x其下游間隔k個位置為y的組合的個數,這樣就P\-k(x,y)表示核苷酸x其下游間隔k個位置為y的條件概率。p(x)和p(y)分別是基因序列中核苷酸x和y的概率。
當選取k=0時,就表示了緊鄰二聯體核苷酸的關聯程度,k=1時表示次緊鄰二聯體核苷酸的關聯程度。[3]i\-k就是基因序列的平均互信息(AMI),不同的k值對應不同的i\-k,對于每一基因組,我們都能夠得到一組數據i\-0,i\-1,…,i\-k,從而構成向量I=
(i\-0,i\-1,…,i\-k),不同的基因序列,可以得到不同的向量I,J,L。
1.2 相關系數
在本文中我們使用的是線性相關系數,它反了映兩個數據集之間的線性相關程度。若相關系數為,表示兩個數據集之間呈現完美的正線性相關;若相關系數為,則表示量數據集之間是負線性相關;若相關系數為0,則表示兩組數據集之間沒有線性相關性。
1.3 聚類分析
我們通過計算不同物種兩兩之間的AMI距離,可以得到不同物種之間的距離從而得到一個距離矩陣。對于這個矩陣,本文選用離差平方和方法進行聚類。
2.基因數據與結果討論
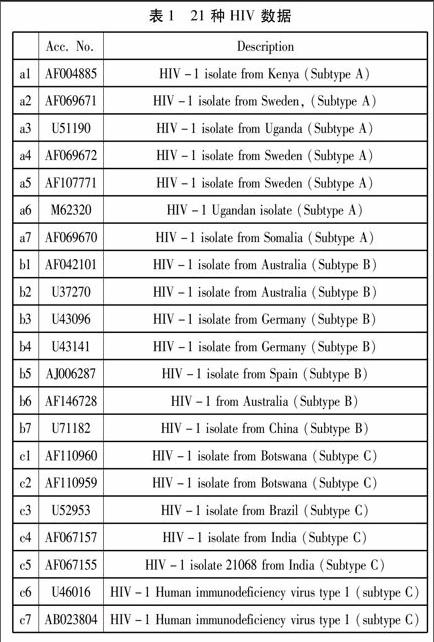
2.1 基因數據:21種HIV 數據
21種HIV病毒基因分為三種亞型,用a、b、c分別表示,每種又各有七種,數據來自NCBI(http://www.ncbi.nlm.nih.gov)。這21種HIV數據,長度比較一致,都在10000個核苷酸上下。
2.2 數據計算
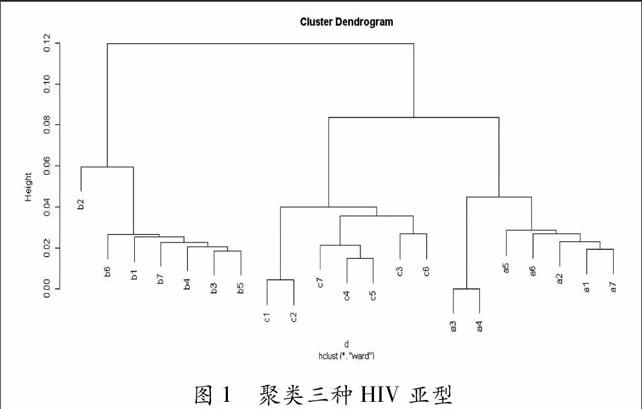
對于這21種HIV病毒,首先提取各自序列的AMI向量,每種病毒得到一個向量 。對于參數k的選取,我們選取了10、50、100、200、300、400、500、600、700等多個值,綜合各向量之間的距離和聚類分析的結果來看,k取500是比較合適的,k過小會丟失基因組的一些關聯信息,k過大對結果沒有什么影響,這樣AMI向量共有501個分量。對于這21個向量計算兩兩之間的相關系數,從而組成一個距離矩陣。我們將這個距離矩陣輸入R軟件,使用離差平方和法進行聚類分析,得到分類結果如圖1所示:
3.結果分析
在Mark等人[4]的文章中,也對這組數據做了分析,Mark等使用UPGMA tree、2維和3維圖等方法對這組數據做了分析,將這21種HIV病毒分成了三類。在本文中我們使用R軟件,應用離差平方和法更為簡單方便,計算速度更快,由圖1可以看出同樣對這21種HIV病毒做了很好的區分,將其分為a、b、c三類,達到完全區分的目的。
[參考文獻]
孫嘯,陸祖宏,謝建明.生物信息學基礎[M].北京:清華大學出版社,2005:238-239.
[2] Mark Bauer,Sheldon M Schuster and Khalid Sayood.The Average Mutual Information Profile as a Genomic Signature[J].BMC Bioinformatics 2008,9:48 doi:10.1186/1471-2105-9-48.
[3] 羅遼復.生命進化的物理觀[M].上海:上海科學技術出版社,2000,168-183.
[4] Ouyang Z,Zhu H,Wang J,et al.Multivariate entropy distance method for p rokaryotic gene identification [J]. J Bioinform ComputBiol, 2004,2(2):353-73.
(作者單位:武警山東省總隊訓練基地,山東 濟南 250000)endprint
