淺析不同含量CoO助劑對(duì)催化劑性能的影響
2015-05-30 11:50:16斯欽德力根等
中小企業(yè)管理與科技·下旬刊 2015年3期
斯欽德力根等
摘要:制備了一系列不同CoO含量相同Mo2C負(fù)載量Mo2C/γ-
Al2O3催化劑。利用微型固定床反應(yīng)器對(duì)其甲烷二氧化碳重整制合成氣反應(yīng)進(jìn)行了活性評(píng)價(jià),并采用XRD方法進(jìn)行了結(jié)構(gòu)表征。當(dāng)在相同負(fù)載量下改變CoO的添加量來(lái)考察其催化劑性能時(shí),結(jié)果表明對(duì)不同含量的CoO添加催化劑來(lái)說(shuō),1%CoO助劑碳化鉬催化劑優(yōu)于5%CoO助劑碳化鉬催化劑。
關(guān)鍵詞:Mo2C/γ-Al2O3催化劑 甲烷二氧化碳重整 合成氣XRD 助劑 CoO
在室溫環(huán)境下,碳化物幾乎耐各種化學(xué)腐蝕,其特點(diǎn)主要表現(xiàn)為:熔點(diǎn)和硬度比較高、熱穩(wěn)定性和機(jī)械穩(wěn)定性極高等。此外,在電、磁性質(zhì)方面,碳化物的金屬屬性與其母體相類似。憑借這些特點(diǎn)和性質(zhì),在機(jī)械切削、礦物開(kāi)采等領(lǐng)域,碳化物得到廣泛的使用[1,2]。
在甲烷制合成氣中,英國(guó)的Edman Tsang最早發(fā)現(xiàn)碳化鉬具有高催化活性,在研究MoO3/SiO2甲烷化轉(zhuǎn)化催化劑的過(guò)程中,Edman Tsang發(fā)現(xiàn),該體系的催化劑,其活性都比較高,合成氣具有驚人的收率。
英國(guó)學(xué)者Anderew York[3]指出,碳化鎢和碳化鉬,對(duì)甲烷轉(zhuǎn)化制合成氣反應(yīng),主要包括蒸氣重整、二氧化碳干氣重整和氧氣重整三種方法,這三種方法都具有較好的催化性能,與大多數(shù)鉬族金屬的催化性能相比,碳化鉬可以相媲美,在一定程度上可以使甲烷轉(zhuǎn)化反應(yīng)達(dá)到熱力學(xué)平衡,提高甲烷化轉(zhuǎn)化率,同時(shí)其合成氣的選擇性超過(guò)90%。
三個(gè)反應(yīng)對(duì)于天然氣的利用及合成染料的生產(chǎn)具有非常重要的意義。甲烷二氧化碳催化重整制合成氣反應(yīng)由于能生產(chǎn)H2/CO比約為1的合成氣,并且這一反應(yīng)對(duì)于環(huán)境保護(hù)與綜合利用資源具有重大意義,而日益受到人們的重視。
本文介紹了γ-Al2O3負(fù)載的碳化鉬在甲烷二氧化碳重整制合成氣反應(yīng)的催化作用,并且分析了相同負(fù)載量下不同含量CoO助劑對(duì)催化劑性能的影響。
1 實(shí)驗(yàn)部分
1.1 催化劑的制備
我們采取的Mo2C/γ-Al2O3催化劑氧化態(tài)前驅(qū)物制備方法是浸漬法,參照Boudart等人提出的程序升溫反應(yīng)方法,制備 Mo2C/γ-Al2O3催化劑[4~5]。
制備氧化鉬前驅(qū)物[6],在500℃[7]環(huán)境下,利用鉬酸銨(A.R.)加熱分解制得,擔(dān)載型氧化鉬前驅(qū)物是用按計(jì)量配好的鉬酸銨(A.R.)-氨水溶液中浸漬γ-氧化鋁[8](粒徑10~50nm,大連路明納米材料有限公司),進(jìn)行0.5h的攪拌,在室溫下放置過(guò)夜,在80~90℃的水浴環(huán)境中蒸發(fā)至干,然后在115℃的烘箱中烘干12h,最后通過(guò)4h500℃焙燒[8]制得。Mo2C和擔(dān)載的Mo2C催化劑是由上面制得的氧化態(tài)前驅(qū)物。將CH4和H2按照1:4的體積比進(jìn)行混合,在混合氣體作為碳化氣的條件下,按照程序升溫碳化制得。選擇內(nèi)徑為4mm的石英玻璃管作為反應(yīng)管進(jìn)行碳化處理,為了支撐樣品,需要在中間塞制石英纖維,氧化態(tài)前驅(qū)物的裝填量控制在0.2g,碳化氣流速控制在35mL/min。實(shí)施碳化時(shí),溫度從室溫升到300℃,升溫速率控制在10℃/min,然后從300℃升到850℃,升溫速率控制在1℃/min,同時(shí)在850℃下恒溫處理2h,接著用流速為28mL/min的氫氣吹掃0.5h,進(jìn)而制得碳化物催化劑。
1.2 催化劑的活性評(píng)價(jià)
碳化之后的碳化物樣品或者原位(insitu)活性評(píng)價(jià),或者在氦氣氛中冷卻到室溫后,用含1(v/v)%O2的N2混合氣體于室溫下鈍化樣品12h,再進(jìn)行結(jié)構(gòu)表征。催化劑活性評(píng)價(jià)在碳化完成并用氫氣(28ml/min)吹掃半小時(shí)之后原位進(jìn)行。甲烷二氧化碳催化重整制合成氣反應(yīng)產(chǎn)物用SP3420型氣相色譜儀(北京分析儀器廠)在線分析,5A分子篩和Porapak-Q填充柱分離,TCD檢測(cè)。
1.3 催化劑的X射線衍射分析
X-射線結(jié)構(gòu)分析是采用德國(guó)D8 Advance型X-射線衍射儀,Cu靶,Ni濾波,Si-Li探測(cè)器,40kV×40mA,掃描范圍:10°~80°,掃描速度:3°/min。
2 結(jié)果與討論
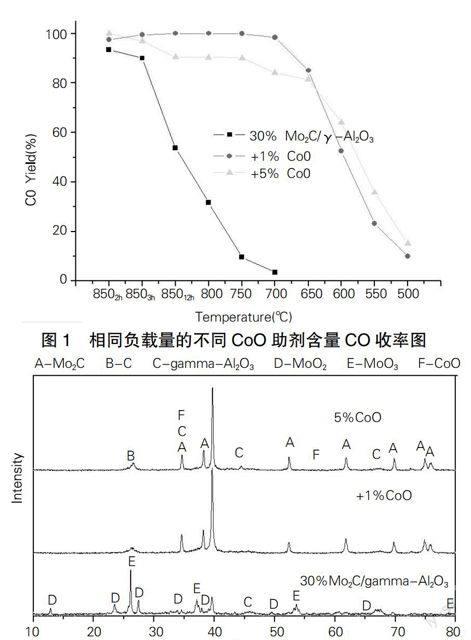
不同含量CoO助劑對(duì)甲烷二氧化碳重整制合成氣反應(yīng)的催化劑Mo2C/γ-Al2O3進(jìn)行對(duì)比,從圖1可以看出加1%CoO助劑催化劑的650℃以上的CO收率高于5%CoO助劑催化劑,加1%CoO助劑催化劑的650℃以下的CO收率低于5%CoO助劑催化劑。總體來(lái)說(shuō)1%CoO助劑碳化鉬催化劑優(yōu)于5%CoO助劑碳化鉬催化劑。圖2可看出1%CoO助劑碳化鉬催化劑X射線衍射峰強(qiáng)于5%CoO助劑碳化鉬催化劑,也就是前者的碳化程度更好,碳化鉬晶型更加完整。所以Co的添加量在一定的范圍之內(nèi)更佳,不是越多越好。
3 結(jié)論
從圖2可看出添加1%CoO助劑的催化劑Mo2C晶型比較完整,氧化態(tài)MoO2、MoO4晶型不明顯,積碳比較少。從而不同含量的CoO添加Mo2C/γ-Al2O3催化劑來(lái)說(shuō)1%CoO助劑催化劑,無(wú)論是CO收率,還是催化劑活性都優(yōu)于5%CoO助劑碳化鉬催化劑。
參考文獻(xiàn):
[1]Oyama S T.The Chemistry of Transition Metal Carbides and Nitrides(ed. Oyama S)Glasgow:Blackie Academic and Professional,1996.1.
[2]Luthin J,Linsmeier C H.J.Nuclear Materials,2001,290 293:121-125.
[3]Andrew York.Shrinking reserves of platinum and other Group Ⅶ tran-sition metals are causing chemists to investigate alternative-catal-ysts using tungsten and molybdenum[J].Chemistry in Britain,1999,35(8).
[4]Blekkan E A,Pham-Huu C,Ledoux M J,etal.Isomerization of n-Heptane on an Oxygen-Modified Molybdenum Carbide Catalyst[J].Ind Eng Chemres,1994,33:1657-1664.
[5]Pham-Huu C,York APE,Benaissa M,etal Reaction of n-Heptane and Methylcyclopentane over an Oxygen-Modified Molybdenum Carbide Catalyst.Study of Coke Formation,Catalyst Deactivation,and Regeneration[J].Ind Eng Chem Res,1995,34:1107-1113.
[6]朱全力,楊建,季生福,等.Mo2C/Al2O3催化劑用于甲烷部分氧化(POM)制合成氣的研究[J].分子催化,2003,17(2):118-123.
[7]朱伯仲,尚雪亞,林鈺,等.利用熱重差熱技術(shù)研究鉬酸銨的熱分解[J].鄭州大學(xué)學(xué)報(bào)(自然科學(xué)版),1997,9(3):71-73.
[8]A.J.Brungs,A.P.E.York,J.B.Claridge.etal.Dry reforming of methane to synthesis gas over supported molybdenum carbide catalysts[J].Catalysis,2000,70:117-122.
作者簡(jiǎn)介:斯欽德力根(1975-),內(nèi)蒙古赤峰人,講師,工學(xué)碩士,研究方向:化學(xué)工藝。
