超高效液相色譜-串聯質譜法測定不同茶葉中草甘膦、氨甲基膦酸及草銨膦的殘留
2015-06-21 12:56:38諸力陳紅平周蘇娟王川丕劉新
分析化學 2015年2期
諸力 陳紅平 周蘇娟 王川丕 劉新
(中國農業科學院茶葉研究所農業部茶葉產品質量安全風險評估實驗室(杭州),杭州310008)
1 引言
草甘膦(Glyphosate)作為無選擇類除草劑,以其高效、低毒、廉價等特性被廣泛運用于全球各個農業和非農業領域[1]。草銨膦(Glufosinate)可以有效去除絕大部分耐草甘膦雜草,且具有環保易降解等特點而受到各界青睞。隨著草甘膦、草銨膦使用頻率不斷上升,特別是在茶園中的運用日益劇增,其殘留問題越來越受到關注,在2013 歐盟農殘標準-茶葉中規定,草甘膦和草銨膦最高殘留限量(MRL)值分別為2.0 和0.1 mg/kg。草甘膦及其主要降解產物氨甲基膦酸(Aminomethyl phosphonic acid,AMPA)和草銨膦之間化學結構相似,都具有易溶于水,難溶于一般有機溶劑,難揮發,缺少發色和熒光基團等特性,因此運用常規方法進行檢測比較困難。
目前,草甘膦檢測方法有色譜法(GC[2]、LC[3,4]、IC[5,6])、分光光度法[7]、質譜法(GC-MS[8]、LC-MSMS[9~11])及電感耦合等離子光譜法(ICP)[12]等,但大部分方法存在靈敏度低、操作程序繁瑣、有機試劑污染嚴重等缺點,也難以直接運用于茶葉農殘檢測領域。
茶葉作為有典型基質效應的經濟作物,其草甘膦和草銨膦檢測文獻報道甚少。食品安全國家標準-食品中農藥最大殘留限量GB2763-2014 規定,茶葉中草甘膦指定檢測方法SN/T 1923-2007 最低定量限為0.1mg/kg,而草銨膦并未指定相應檢測標準。本實驗通過“堿提酸沉”pH 值調控及Oasis HLB 小柱凈化后FMOC-Cl 衍生前處理,采用可編程多反應監測(Scheduled MRM),建立了一種超高效液相色譜-串聯質譜測定不同茶葉(綠茶、紅茶、烏龍茶、普洱茶)中草甘膦、氨甲基膦酸及草銨膦的方法。本方法穩定、簡便、靈敏,可滿足各類茶葉檢測需求。
2 實驗部分
2.1 儀器與試劑
API 3200 型串聯三重四極桿質譜(美國AB 公司);Waters ACQUITY 超高效液相色譜儀(Waters 公司);Mill-Q 去離子水發生器(美國Millipore 公司);Sigma 3-16L 離心機(德國Sigma 公司);Oasis HLB固相萃取柱(3 mL/60 mg,Waters 公司)。
草甘膦、草銨膦、氨甲基膦酸標準樣品(純度≥98.0%,德國Dr.Ehrenstorfer 公司);氯甲酸-9-芴基甲酯(FMOC-Cl,不低于98.0%,百靈威公司);甲醇、乙腈(色譜純,百靈威公司);甲酸銨(99.9%,德國Sigma 公司);甲酸(95%,德國Sigma 公司);KOH(分析純,杭州蕭山化學試劑廠);HCl(36% ~38%,優級純,江蘇永華精細化學品有限公司);硼酸鈉(Na2B4O7·10H2O,99.5%,太倉美達試劑有限公司);5%(V/V)硼酸鹽緩沖溶液(pH=9)。
茶葉樣品來源于浙江省各茶葉市場及各大超市。
2.2 色譜和質譜條件
色譜柱:Waters ACQUITY HSS T3 (100 mm × 2.1 mm,1.8 μm);流動相:含0.1%甲酸-1 mmol/L甲酸銨溶液(流動相A)和甲醇溶液(流動相B);梯度洗脫程序:0 ~1 min,10% B;1 ~6 min,10% ~100% B;6 ~6.1 min,100% ~10% B;6.1 ~7.0 min,10% B。流速:0.3 mL/min;進樣量:10.0 μL;柱溫:40 ℃;運行時間:7 min。
離子源:電噴霧離子源(ESI),溫度500 ℃,電壓5500 V;掃描方式:正離子模式;霧化氣GS1 壓力:50 psi;霧化氣GS2 壓力:50 psi;定性與定量離子對、去簇電壓(DP)、碰撞能量(CE)等參數見表1。
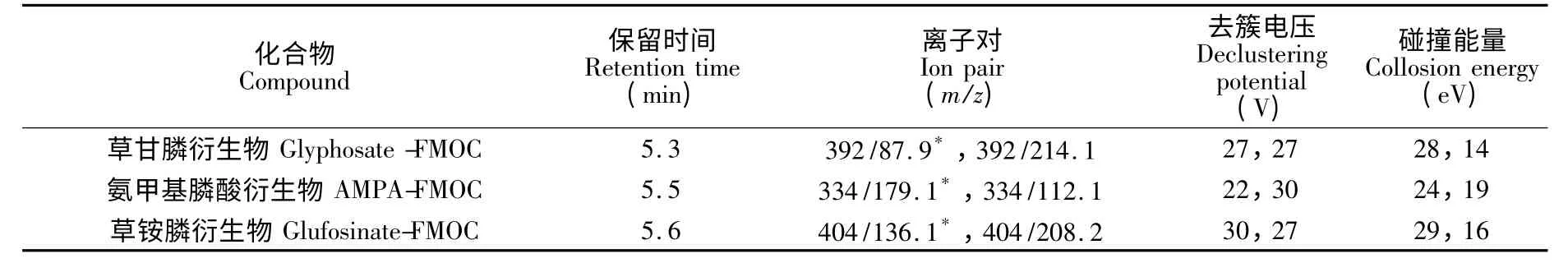
表1 草甘膦、草銨膦及氨甲基膦酸衍生物質譜條件參數Table 1 MS conditions for derivatizations of glyphosate,glufosinate and aminomethyl phosphonic acid (AMPA)
2.3 樣品制備
提取:稱取2.0 g 茶葉試樣于50mL 離心管,加入0.05 mol/L KOH 溶液15 mL,搖勻靜置20 min。經5000 r/min 離心10 min,過濾,取濾液2 mL 至5 mL 離心管,加入5 mol/L HCl 溶液20 μL,振蕩均勻后經5000 r/min 離心10 min,得到上清液。
凈化:取1 mL 上清液至Oasis HLB(3 mL/60 mg)小柱(小柱活化:先加2 mL 甲醇淋洗,再加2 mL水活化),經過柱后,用5 mL 離心管接取凈化流出液。
衍生:凈化液中加入0.2 mL 硼酸鈉緩沖溶液,搖勻后再加入0.1 mL 10 g/L FMOC-Cl 衍生試劑,振蕩均勻后靜置2 h。將衍生后液體試樣10000 r/min 離心10 min,經0.22 μm 水系濾膜,供液相色譜-串聯質譜分析。
2.4 標準曲線制作
分別稱取3 種標準品各0.05 g,用水溶解并定容至50 mL 塑料容量瓶,分別配制成1000 mg/L 標準溶液,4 ℃下保存待用。
標準曲線的配制:采用空白茶葉(綠茶、紅茶、烏龍茶、普洱茶),經2.3 節提取凈化,分別配制成6 個不同濃度濃度(5 ~1000 μg/L)的基質標樣,現配現用。
2.5 標準加入回收實驗
稱取不同茶葉空白樣2 g,分別添加當量為0.1、0.4 和4.0 mg/kg 混合標準溶液,每個濃度水平重復6 次,經2.3 節提取凈化處理,采用各茶葉基質標準曲線進行外標法定量,計算回收率、相對標準偏差和定量限。
3 結果與討論
3.1 質譜和色譜條件優化
分別取草甘膦、草銨膦及氨甲基膦酸濃度為8 mg/L 標樣進行衍生,用于衍生化合物質譜條件優化。本研究采用ESI+模式,選擇各準分子離子[M +H]+作為母離子,進行二級質譜優化,參數包括電噴霧電壓、碰撞電壓(CE)、去簇電壓(DP)、電離溫度等,優化結果見表1。采用可編程多反應監測(Sched-uled MRM,SMRM),該模式可以不分時間段同時進行多種殘留農藥檢測,極大提高了MRM 檢測通量,有效改善峰形,提高重現性[13]。
研究表明,3 種衍生化合物在HSS T3 柱中的保留強度優于普通C18柱,故予以采用。流動相優化包括甲醇-水、乙腈-水、甲醇-水(0.1%甲酸和1 mmol/L 甲酸銨)、乙腈-水(0.1%甲酸和1 mmol/L 甲酸銨)。結果表明,采用甲醇-水(0.1%甲酸和1 mmol/L 甲酸銨)作為流動相可提高目標化合物離子化效率,緩沖體系進一步增加系統穩定性,3 種化合物分離度和靈敏度都達到最佳。采用2.2 節梯度程序洗脫,不斷增加流動相中甲醇的比例,有助于降低基質對目標化合物檢測干擾,同時可有效減少雜質對色譜柱的污染[14]。圖1 為0.04 mol/L 綠茶基質標準溶液(A)、空白綠茶樣品溶液(B)、加標0.4 mg/kg 綠茶樣品溶液(C)中3 種衍生化合物的二級質譜總離子流色譜圖。由圖1 可見,分離效果良好,目標化合物出峰段雜質干擾較小。

圖1 0.04 mol/L 綠茶基質標準溶液(A)、空白綠茶樣品溶液(B)、添標0.4 mg/kg 綠茶樣品溶液(C)中3 種衍生化合物的二級質譜總離子流圖Fig.1 MS/MS total ion chromatograms of derived compounds in matrix standard solution (A),blank solution (B)and 0.4 mg/kg spiked concentration levels (C)of green tea
3.2 前處理條件優化
草甘膦、草銨膦和氨甲基膦酸都屬于強極性化合物,通常采用純水或水/二氯甲烷混合作為提取溶劑[15]。由于茶葉富含色素、糖、氨基酸、多酚類等大量水溶性雜質,采用純水提取時,會導致提取液成分復雜,目標化合物離子化效率過低[16],方法定量限難以滿足檢測要求。本實驗經比較后選擇強堿溶液,主要原因是:在堿性環境下,弱酸性草甘膦等會反應生成相應鹽,可提高其溶解率,而且茶多酚更易氧化生成沉淀,達到初步凈化目的。
在提取率研究方面,本研究將預先篩選的陽性綠茶樣品A(含草甘膦和氨甲基膦酸)和B(含草銨膦)作為提取實驗試樣。以下各分析均設定以最理想條件下所測定值(Relative detection value)為1 作為衡量標準。對樣品A 和B 分別進行0 ~10 mol/L 不同濃度KOH 溶液進行提取,并且添加相應濃度HCl 溶液,用以調節pH 值至中性而不影響衍生效率。如圖2 所示,采用純水作為提取溶液,效果不理想,A和B 樣品中3 種化合物草甘膦、草銨膦、氨甲基膦酸測定結果僅為0.31,0.42,0.38;0.05 mol/L KOH 提取效果最優。
利用HCl 調節pH 值,按照2.3 節提取,離心后取上清液2 mL,分別添加0 ~5 mol/L HCl 溶液0.1 mL,調至中性。如圖2 所示,在HCl 濃度為1 mol/L 時,實測值最高,而未添加HCl 溶液組測定結果為0.56,0.65,0.63。造成該結果原因可能是:HCl 打破了原有茶湯中的電解質平衡,在離子效應、電解質作用和共沉效應等共同作用下,茶多酚、氨基酸、糖類等聚合成大分子物質形成絡合物而沉淀[17],凈化效果提高。

圖2 不同濃度KOH 提取(a)和不同濃度HCl 調控(b)對3 種化合物相對測定值的影響Fig.2 Detectable results of three compounds (a)extracted by different concentrations of KOH and(b)regulated by different concentrations of HCl
本研究選擇Oasis HLB(3 mL/60 mg)小柱,其填料為親水親脂的反相吸附劑,由N-乙烯吡咯烷酮和親脂性二乙烯基苯聚合而成,已在抗生素[18]、激素[19]等檢測領域廣泛運用。分別將50 和500 μg/L的3 種化合物溶劑標樣通過HLB 小柱凈化,測得回收率在92.3% ~106.9%之間,說明該小柱并未對3 種化合物產生吸附,因此本方法直接接取凈化流出液進行衍生[9],省去傳統洗脫、濃縮等步驟,縮短操作流程。通過測定過柱前后茶葉提取液發現,該過程中去除了95.6%茶多酚、90.4%咖啡堿及53.2%氨基酸,證明凈化效果良好。
FMOC-Cl 衍生機制[11,20]是在堿性環境(pH=9)下通過FOMC-基團取代目標化合物氮原子上的氫,從而生成較穩定的化合物FOMC-R。在不同茶葉基質中添加過量衍生試劑條件下,對衍生反應時間、溫度及pH 值進行優化,結果表明,反應2 h 后,測得衍生化合物無明顯增加,改變溫度對衍生率影響不大,pH=9 時衍生率最佳。
3.3 線性范圍、相關系數和基質效應評價
采用2.4 節中制備的各基質混合標準溶液,按優化后檢測條件進樣,以各定量離子對峰面積對應濃度做標準曲線,得到3 種化合物線性方程及其相關系數。由表2 可見,3 種化合物在5 ~1000 μg/L 濃度范圍內線性良好(R2>0.99)。草甘膦和草銨膦衍生物在不同基質中基質效應(Matrix effect,Me)不明顯,范圍在89.7% ~103.3%之間。氨甲基磷酸衍生物在綠茶中無明顯基質效應,但在烏龍茶和普洱茶中Me 分別為73.8%和76.4%,其中基質抑制最強為紅茶,Me 達60.2%,原因可能是茶葉經不同程度發酵產生的色素等會對目標化合物測定產生影響。為消除基質效應干擾,本研究采用相應基質匹配標樣進行校準。

表2 不同茶葉基質中3 種化合物的線性方程、相關系數及添標回收率、精密度和方法定量限(n=6)Table 2 Linear equations,correlation coefficients,average recoveries,relative standard deviation(RSD),and limit of quantification(LOQ)of three compound in different matrix(n=6)
3.4 回收率、精密度和方法定量限
取不同茶葉2 g,分別添加0.1,0.4 和4 mg/kg 混合標樣,按照前述方法處理平行測定6 次,回收率和精密度見表2。不同茶葉基質中3 種化合物添標回收率范圍在72.1% ~109.9%之間,RSD(n =6)在0.5% ~9.8%之間,表明所建方法重復性良好。以10 倍信噪比(S/N)計算方法定量限(LOQ)在0.03 ~0.08 mg/kg 之間。
3.5 實際樣品分析
運用此方法對市場247 份樣品進行測試,其中綠茶146 份、紅茶44 份、烏龍茶42 份、普洱茶15 份。測試結果,草甘膦和草銨膦檢出樣品分別為46 份和4 份,目標物含量范圍為0.105 ~3.223 mg/kg 和0.124 ~1.351 mg/kg(統一檢出限為0.1 mg/kg)。本方法可滿足歐盟對茶葉制定農藥最高殘留限量(MRLs)要求,為茶葉行業中制定相關檢測標準提供了理論依據。
1 SU Shao-Quan.Chinese J.Pest.,2005,44(4):145 -149
蘇少泉.農藥,2005,44(4):145 -149
2 Hu J Y,Chen C L,Li J Z.J.Anal.Chem.,2008,63(4):371 -375
3 ZHI Jian-Liang,MOU Ren-Xiang,CHEN Ming-Xue,ZHU Zhi-Wei.Chinese J.Anal.Lab.,2008,27:170 -172
支建梁,牟仁祥,陳銘學,朱智偉.分析試驗室,2008,27:170 -172
4 FANG Fang,XU Hui,WEI Rong-Qing,LIU Xiao-Ning,XU Rong,LI Shou-Chun,LIU Bao-Kun.Journal of Instrumental Analysis,2011,30(6):683 -686
方芳,徐會,魏榮卿,劉曉寧,徐蓉,李壽椿,劉寶菎.分析測試學報,2011,30(6):683 -686
5 YE Li-Min,HU Zhong-Yang,PAN Guang-Wen.Chinese J.Anal.Chem.,2011,39(11):1762 -1765
葉立民,胡忠陽,潘廣文.分析化學,2011,39(11):1762 -1765
6 QIU Hui-Min,GENG Jin-Jü,HAN Chao,REN Hong-Qiang.Chinese J.Anal.Chem.,2013,41(12):1910 -1914
邱慧敏,耿金菊,韓超,任洪強.分析化學,2013,41(12):1910 -1914
7 DONG Wen-Geng,CHEN Xue-Cheng,LANG Zhi-Min,HE Sen,YING Jian-Qun.Chinese J.Anal.Chem.,1997,25(10):1210 -1212
董文庚,陳學誠,朗志敏,何森,英建群.分析化學,1997,25(10):1210 -1212
8 Anastassiades M,Lehotay S J,Tajnbaher D,Schenck F J.J.AOAC Int.,2003,86:412 -431
9 Yoshioka N,Asano M,Kuse A,Mitsuhashi T,Nagasaki Y,Ueno Y.J.Chromatogr.A,2011,1218(23):3675 -3680
10 Ibanez M,Pozo O J,Sancho J V,Lopez F J,Hernandez F.J.Chromatogr.A,2005,1081(2):145 -155
11 Botero-Coy A M,Ibanez M,Sancho J V,Hernandez F.J.Chromatogr.A,2013,1292 :132 -141
12 Guo Z X,Cai Q T,Yang Z G.J.Chromatogr.A,2005,1100:160 -169
13 Zhang K,Wong J W,Yang P,Hayward D G,Sakuma T,Zou Y Y,Schreiber A,Borton C,Nguyen T V,Kaushik B,Oulkar D.J.Anal.Chem.,2012,84(13):5677 -5684
14 ZHANG Xin-Zhong,LUO Feng-Jian,LIU Guang-Ming,LOU Zheng-Yun,CHEN Zong-Mao.Chinese J.Anal.Chem.,2011,39(9):1329 -1335
張新忠,羅逢健,劉光明,樓正云,陳宗懋.分析化學,2011,39(9):1329 -1335
15 Chen M X,Cao Z Y,Jiang Y,Zhu Z W.J.Chromatogr.A,2013,1272:90 -99
16 Kittlaus S,Schimanke J,Kempe G,Speer K.J.Chromatogr.A,2011,1218(46):8399 -8410
17 HUANG Hui-Hua,WANG Shao-Bin,WANG Zhi,LIANG Han-Hua.Food Sci.,2002,23(1):26 -30
黃惠華,王少斌,王志,梁漢華.食品科學,2002,23(1):26 -30
18 ZHOU Ai-Xia,SU Xiao-Si,GAO Song,ZHANG Yu-Ling,LIN Xue-Jue,ZHANG Lan-Ying,An Yong-Lei.Chinese J.Anal.Chem.,2014,42(3):397 -402
周愛霞,蘇小四,高松,張玉玲,林學玨,張蘭英,安永磊.分析化學,2014,42(3):397 -402
19 LAI Shi-Yun,TAO Bao-Hua,FU Shi-Shan,HE Guang-Hua,WEI Ying,ZHANG Shun-Jing,REN Yi-Ping.Chinese J.Anal.Chem.,2012,40(1):135 -139
賴世云,陶保華,傅士姍,何光華,魏瑩,張京順,任一平.分析化學,2012,40(1):135 -139
20 Ghanem A,Bados P,Kerhoas L,Dubroca J,Einhorn J.J.Anal.Chem.,2007,79(10):3794 -3801
