基于高通量測序技術檢測全基因組DNA甲基化水平的方法
2015-10-27 07:17:36馬浪浪梁振娟先新羅仕文李清超梁黔云
生物技術通報 2015年7期
馬浪浪梁振娟先新羅仕文李清超梁黔云
(1. 四川農業大學玉米研究所,成都 611130;2. 畢節市農業科學研究所,畢節 551700)
基于高通量測序技術檢測全基因組DNA甲基化水平的方法
馬浪浪1,2梁振娟2先新2羅仕文2李清超2梁黔云2
(1. 四川農業大學玉米研究所,成都 611130;2. 畢節市農業科學研究所,畢節 551700)
DNA甲基化的研究最近幾年一直是表觀遺傳學研究的重點。有關DNA甲基化水平檢測的方法大體上有十幾種,隨著第二代測序技術的發展,實現了在全基因組水平上對甲基化狀態進行檢測。目前,基于第二代高通量測序進行全基因組DNA甲基化水平檢測的方法主要有BSP-seq(亞硫酸氫鹽修飾結合直接測序法)、MeDIP-seq(甲基化DNA免疫共沉淀測序法)、MBD-seq(甲基化DNA富集結合高通量測序法)。就這3種方法在原理、流程、優缺點、優化使用及后期需要用到的部分生物信息學資源等方面的研究進展作一綜述,旨在為研究者在采用高通量測序方法研究DNA甲基化模式時提供一些思路。
高通量測序技術;全基因組;DNA甲基化水平;BSP-seq;MeDIP-seq;MBD-seq
表觀遺傳學的研究日益深入,作為其中重要一方面的DNA甲基化更是長期研究的熱點。人們越來越清楚地認識到有關DNA甲基化的研究很有可能在醫學、農學等領域產生重大變革。
1 DNA甲基化的基本特征及維持機制
DNA甲基化是指S-腺苷甲硫氨酸上的甲基在甲基化轉移酶的作用下轉移到DNA胞嘧啶或腺嘌呤上,從而對DNA進行修飾,進而發生一系列的表觀修飾現象[1]。
與其他研究相似,人們對于植物DNA甲基化修飾的研究始于模式植物擬南芥,擬南芥全基因組甲基化圖譜(單核苷酸分辨率)已經公布[2-4]。研究表明,在植物中發生DNA甲基化修飾的區域主要有CpG島、CHH、CHG(H代表A或T)以及轉錄區域、基因主體上[2-7]。發生于CpG島、CHH、CHG區域的DNA甲基化修飾主要生物學功能為:通過產生甲基化重復序列來抑制基因組DNA,從而達到保護基因組的目的;發生于轉錄區域、基因主體區域的DNA甲基化修飾主要生物學功能為:通過甲基化水平的高低調節基因轉錄水平。一系列研究都表明,基因的甲基化水平與轉錄水平呈負相關性[4,6,8]。
DNA甲基化能引起一系列表觀遺傳修飾現象:植物開花、衰老、抗逆、基因沉默及雜種優勢等。如Sheldon等[9]研究表明抽薹相關基因的甲基化水平降低,可以造成植物提取開花。Fraga等[10]研究發現成年輻射松植株的甲基化程度明顯高于幼年植株的甲基化程度。Roberta等[11]對三葉草根系、大麻根系進行重金屬處理后發現它們的基因組DNA甲基化水平均降低。Peerbolte等[12]研究表明在T-DNA上的冠癭堿基因發生甲基化作用,使得該基因在隨后的轉化過程中沉默。孫其信[13]對植物分別進行自交、雜交,結果表明自交后基因組DNA甲基化水平增加,雜交后基因組DNA甲基化水平降低。本課題組以玉米幼胚胚性愈傷組織誘導過程為切入點,對該過程中不同時期全基因組DNA甲基化模式進行了分析(運用了甲基化DNA免疫共沉淀結合高通量測序技術,即MeDIP-seq,該技術將會在后文闡述),得出了在這一過程中DNA甲基化水平的變化情況,具體研究結果待發表。以上幾個例子表明:在一系列的生物學過程中都有DNA甲基化的參與,隨著研究方法、技術的提高,研究者對于DNA甲基化參與表觀遺傳修飾的研究必將更加深入。
目前,有關DNA甲基化的維持機制主要基于4類甲基轉移酶進行作用。如MET1甲基轉移酶在DNA復制過程中能識別單拷貝及重復序列的CG位點,使得在該CG位點發生甲基化作用[14,15];DRM重新甲基化酶則是在小分子RNA介導下識別胞嘧啶序列,進而發生重新甲基化作用,即RdDM[16,17];CMT染色質甲基化酶可以特異性作用于異染色質區的CNG序列,進而發生甲基化作用,該酶在植物中特有存在[18-20];研究者認為DNMT2家族的一些同系物可能也參與甲基化的維持機制,具體方式有待進一步研究[21]。這幾類甲基轉移酶有時也并非獨立參與機制的調控,它們常常相互作用,共同參與機制的調控。我們相信,隨著研究技術的不斷提升,研究者對于DNA甲基化的維持機制將會有更加全面的認識。
2 基于全基因組進行DNA甲基化檢測的方法
目前,研究者對于DNA甲基化的檢測方法主要可以分為兩個層面:(1)基于全基因組DNA甲基化水平檢測;(2)基于片段(位點)DNA甲基化檢測。基于全基因組DNA甲基化的檢測方法,見表1。
上述基于基因組DNA甲基化的研究方法中,BSP-seq、MeDIP-seq和MBD-seq這3種方法隨著全基因組高通量測序技術的發展以及生物信息學的興起,在人們的研究中越來越受到重視。故下文主要針對這3種方法進行介紹。
2.1 三種檢測方法的工作流程
2.1.1 BSP-seq法檢測DNA甲基化水平 BSP-seq法最早是由Frommer等[28]提出,其檢測DNA甲基化的原理已在表1中介紹。該方法的檢測流程如下:(1)基因組DNA的提取、檢測、純化(保證DNA的質量、純度等);(2)用亞硫酸氫鈉處理基因組DNA;(3)對修飾后的DNA進行回收并純化;(4)引物設計(根據目的基因啟動子序列);(5)PCR擴增目的片段;(6)PCR產物克隆、測序;(7)對所得序列進行比對、對所得數據進行生物信息學分析[28,31]。
運用BSP-seq檢測DNA甲基化需要注意的問題:(1)樣品的質量應適中,DNA量太多會造成亞硫酸鹽修飾不完全,DNA量太少則會造成后期實驗回收量不足;(2)在亞硫酸鹽修飾過程中必須保證pH值絕對準確、且需要多次摸索合適的反應時間;(3)因為該過程涉及到PCR擴增,故引物設計、PCR反應體系都需不斷優化[32]。此外,在后期數據處理時可能會碰到一些問題,如由于測序錯誤、污染等情況,得到的數據處理時比較麻煩。針對這些情況,Krueger等[33]給出一些處理方法,使得實驗結果的準確性提高。
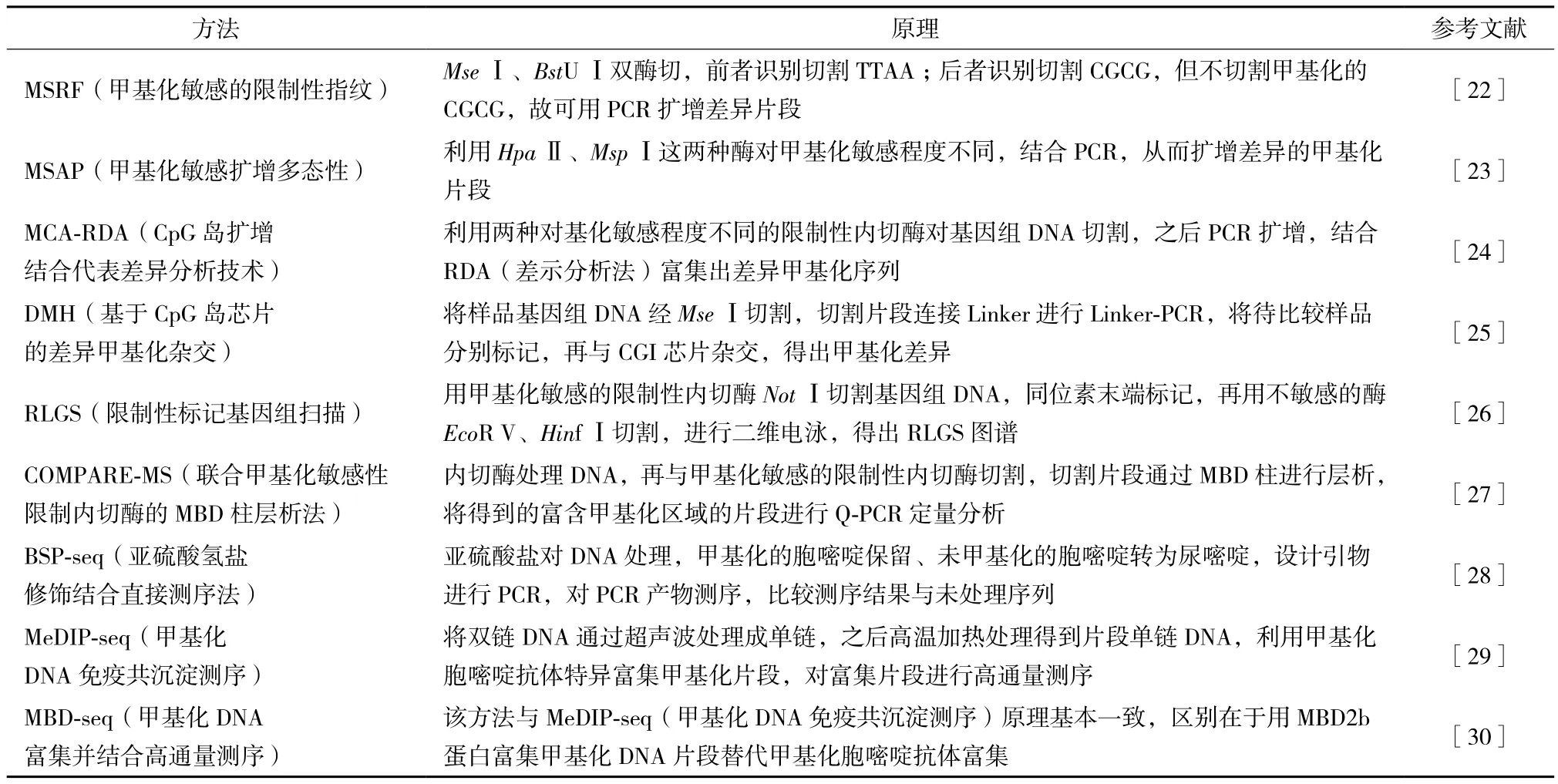
表1 基于全基因組進行DNA甲基化的檢測方法
2.1.2 MeDIP-seq法檢測DNA甲基化水平 MeDIP-seq法是由Weber等[34]在2005年首先提出,是指5'-甲基胞嘧啶抗體特異富集基因組DNA上發生甲基化的片段,之后通過對這些富集的甲基化片段進行高通量測序,從而獲得基因組DNA的甲基化發生區域、甲基化水平等。具體操作流程為:(1)通過超聲波將DNA打斷成DNA片段;(2)通過高溫使得片段DNA解為單鏈;(3)片段DNA與5'-甲基胞嘧啶抗體特異反應,從而構建甲基化DNA文庫(這一過程伴隨著對片段DNA進行PCR等處理);(4)高通量測序及后期對數據進行生物信息學分析(測序數據去污染、去接頭、統計數據產出量;測序序列與參考基因組序列比較;測序數據在基因組的分布;測序數據富集區(Peak區)的信息統計;基于Peak進行全基因組甲基化差異分析等)[29,34]。運用該方法進行基因組DNA甲基化檢測需要注意:(1)DNA片段富集過程中富集條件必須摸索才能達到好的效果;(2)DNA片段富集后,當富集量較少時需采用PCR擴增,PCR反應體系需不斷優化;(3)要保證樣品在整個測序過程中無污染等[35]。
2.1.3 MBD-seq法檢測DNA甲基化水平 MBD-seq法檢測基因組DNA甲基化水平的原理與MeDIP-seq法基本相似。其檢測流程如下:(1)特異性結合甲基化DNA的蛋白MBD2b富集高甲基化的DNA片段;(2)對富集的DNA片段進行末端補平得到平末端片段;(3)對平末端片段進行末端加A',得到3'-dA黏性末端;(4)加連接頭,獲得帶接頭片段,進而得到長度合適的目的DNA片段;(5)進行PCR擴增,構建MBD-seqDNA文庫;(6)進行高通量測序,并對所得結果進行生物信息學分析[30]。
近年,研究者采用該方法對DNA甲基化模式進行了大量的研究,如Wang等[36]采用MBD-seq法分析家族腺瘤性息肉癥患者在BRB栓劑的治療下的甲基化狀態顯示,在該栓劑的作用下出現較多的脫甲基轉錄位點;家族腺瘤性息肉癥患者的息肉減少。綜合該結果表明,家族腺瘤性息肉癥可能與脫甲基化轉錄相關。運用該方法進行基因組DNA甲基化水平檢測時,需要注意的問題同運用MeDIP-seq法需要注意的問題基本相似。
2.2 三種檢測方法的比較及優化使用
BSP-seq、MeDIP-seq、MBD-seq這3種基于高通量測序檢測全基因組DNA甲基化方法在費用、分辨率、數據分析等方面有一定的差異。表2是就BSP-seq、MeDIP-seq和MBD-seq這3種方法在這些方面的差異進行的總結。
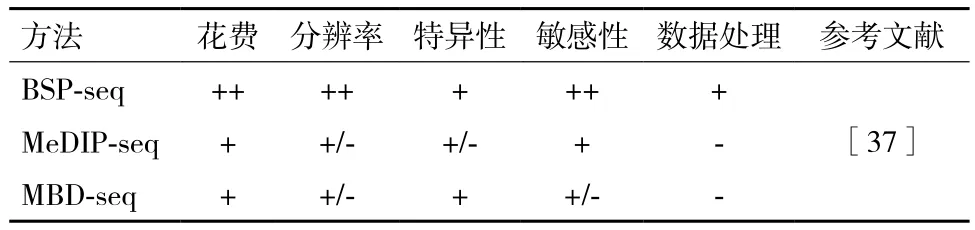
表2 三種基于高通量測序檢測全基因組DNA甲基化方法比較
由于BSP-seq法既可得到全基因組上的甲基化信息,又可得到特異位點上的甲基化信息,故此法被認為是金標準。但對于基因組較大的物種而言,該方法花費巨大,且高通量測序數據也很龐大,對于后期數據處理、分析提出了挑戰。因此,研究者在實際操作中往往很難接受或采用該方法。MeDIP-seq法及MBD-seq法測序費用相對較低,數據量較小,但分析得到的結果很難驗證。基于上述原因,科研工作者進行了大量的改進嘗試。如Li等[38]采用BSP-seq法得出人類外周血單核細胞中的一個單堿基分辨率甲基化組,并以此圖譜作為參考基因組對比了MeDIP-seq法及MBD-seq法的實驗結果,結果顯示,MeDIP-seq對高度甲基化、高CpG密度更加敏感;MDB-seq對高度甲基化、中等CpG密度更加敏感。因此,這兩種方法可以形成很好的互補效應。
2.3 三種檢測方法所涉及的生物信息學分析資源BSP-seq、MeDIP-seq、MBD-seq這3種方法在經過高通量測序后得到大量的數據。對于這些數據需要進行進一步的生物信息學分析,才能得到研究者理想的結果,詳見表3。

表3 部分生物信息學分析資源
3 展望
隨著第二代高通量測序的發展,人們將在越來越多的領域中使用它。DNA甲基化作為表觀遺傳學中重要的一個方面,在近幾年的研究中借助第二代高通量測序技術取得了長足的進展。采用高通量測序技術研究DNA甲基化模式將是一種趨勢。
BSP-seq、MeDIP-seq、MBD-seq是目前基于高通量測序技術研究全基因組DNA甲基化模式的3種方法。正如前文所述,這3種方法各有優劣,結合花費、分辨率、特異性、敏感度及數據分析等方面研究者認為[40]:在針對一些基因組較大的物種進行全基因組DNA甲基化模式研究中采用MeDIP-seq法和MBD-seq法相互補充較優于采用BSP-seq法;而在針對一些基因組較小的物種進行研究時,可以根據研究目標選擇不同的方法(如需要得到高分辨率、高特異性及高敏感度等較高要求時可優先選擇BSP-seq法,反之可選擇MeDIP-seq法或MBD-seq法)。
本課題組采用MeDIP-seq法研究了高胚誘導率玉米骨干自交系幼胚發育過程中全基因組DNA甲基化模式,結合近幾年擬南芥全基因組DNA甲基化已公布圖譜,筆者以為,應以此為模式并結合優化使用BSP-seq、MeDIP-seq、MBD-seq這3種方法開展相關植物全基因組DNA甲基化研究,相信在不久的將來有關DNA甲基化的確切機制將會被人類解析。
[1] Bird AP. CpG-rich islands and the function of DNA methylation[J]. Nature, 1985, 321(6067):209-213.
[2]Cokus SJ, Feng S, Zhang X, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning[J]. Nature, 2008, 452(7184):215-219.
[3]Lister R, O’Malley RC, Tonti-Filippini J, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis[J]. Cell, 2008, 133(3):523-536.
[4]Zhang X, Yazaki J, Sundaresan A, et al. Genome-wide highresolution mapping and functional analysis of dna methylation in Arabidopsis[J]. Cell, 2006, 126(6):1189-1201.
[5]Tran RK, Henikoff JG, Zilberman D, et al. DNA Methylation profiling identifies CG methylation clusters in Arabidopsis genes[J]. Current Biology, 2005, 15(2):154-159.
[6]Zilberman D, Gehring M, Tran RK, et al. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription[J]. Nature Genetics, 2006,39(1):61-69.
[7]Hsieh TF, Ibarra CA, Silva P, et al. Genome-wide demethylation of Arabidopsis endosperm[J]. Science, 2009, 324:1451-1454.
[8]Zemach A, McDaniel IE, Silva P, et al. Genome-wide evolutionary analysis of eukaryotic DNA methylation[J]. Science, 2010, 328(5980):916-919.
[9] Sheldon CC, Finnegan EJ, Rouse DT, et al. The control of flowering by vernalization[J]. Curr Opin Plant Biol, 2000, 3(5):418-422.
[10]Fraga MF, Ca?al M, Rodríguez R. Phase-change related epigenetic and physiological changes in Pinus radiata D. Don[J]. Planta,2002, 215(4):672-678.
[11]Aina R, Sgorbati S, Santagostino A, et al. Specific hypomethylation of DNA is induced by heavy metals in white clover and industrial hemp[J]. Physiologia Plantarum, 2004, 121(3):472-480.
[12]Peerbolte R, Leenhouts K, Hooykaas-van Slogteren GM, et al. Clones from a shooty tobacco crown gall tumor II:irregular T-DNA structures and organization, T-DNA methylation and conditional expression of opine genes[J]. Plant Mol Biol, 1986, 7:285-299.
[13]孫其信. 農作物雜種優勢機理研究及展望[J]. 作物雜志,1998, 4:31.
[14] Bernstein BE, Meissner A, Lander ES. The mammalian epigenome[J]. Cell, 2007, 128(4):669-681.
[15] Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases[J]. Annu Rev Biochem, 2005, 74:481-514.
[16] Chan SW, Zilberman D, Xie Z, et al. RNA silencing genes control de novo DNA methylation[J]. Science, 2004, 303(5662):1336-1336.
[17]Cao X, Aufsatz W, Zilberman D, et al. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation[J]. Current Biology, 2003, 13(24):2212-2217.
[18] Papa CM, Springer NM, Muszynski MG, et al. Maize chromomethylase Zea methyltransferase2 is required for CpNpG methylation[J]. The Plant Cell Online, 2001, 13(8):1919-1928.
[19] Bartee L, Malagnac F, Bender J. Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene[J]. Genes & Development, 2001, 15(14):1753-1758.
[20] Lindroth AM, Cao X, Jackson JP, et al. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation[J]. Science, 2001, 292(5524):2077-2080.
[21]Goodrich J, Tweedie S. Remembrance of things past:chromatin remodeling in plant development[J]. Annual Review of Cell and Developmental Biology, 2002, 18(1):707-746.
[22]Huang THM, Laux DE, Hamlin BC, et al. Identification of DNA methylation markers for human breast carcinomas using the methylation-sensitive restriction fingerprinting technique[J]. Cancer Research, 1997, 57(6):1030-1034.
[23]Reyna-Lopez GE, Simpson J, Ruiz-Herrera J. Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms[J]. Molecular and General Genetics MGG, 1997, 253(6):703-710.
[24]Toyota M, Ho C, Ahuja N, et al. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification[J]. Cancer Res, 1999, 59:2307-2312.
[25]朱燕. DNA 甲基化的分析與狀態檢測[J]. 現代預防醫學,2006, 32(9):1070-1073.
[26] Hatada I, Hayashizaki Y, Hirotsune S, et al. A genomic scanningmethod for higher organisms using restriction sites as landmarks[J]. Proc Natl Acad Sci USA, 1991, 88(21):9523-9527.
[27] Yegnasubramanian S, Lin X, Haffner MC, et al. Combination of methylated-DNA precipitation and methylation-sensitive restriction enzymes(COMPARE-MS)for the rapid, sensitive and quantitative detection of DNA methylation[J]. Nucleic Acids Research, 2006, 34(3):e19.
[28] Frommer M, McDonald LE, Millar DS, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands[J]. Proc Natl Acad Sci USA, 1992,89:1827-1831.
[29] Thu KL, Vucic EA, Kennett JY, et al. Methylated DNA immunoprecipitation[J]. J Vis Exp:JoVE, 2009(23):935.
[30] Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome[J]. Nucleic Acids Research,2010, 38(2):391-399.
[31] Beck S, Rakyan VK. The methylome:approaches for global DNA methylation profiling[J]. Trends Genet, 2008, 24(5):231-237.
[32]http://www. biovol. net/mutation/12852. htm. 上海拜沃生物科技有限公司分子生物學技術服務, 2013-1-29
[33]Krueger F, Kreck B, Franke A, et al. DNA methylome analysis using short bisulfite sequencing data[J]. Nature Methods, 2012,9(2):145-151.
[34]Weber M, Davies JJ, Wittig D, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells[J]. Nature Genetics, 2005, 37(8):853-862.
[35]http://www. plob. org/2012/01/09/1614. html.
[36]Wang LS, Burke C, Hasson H, et al. A phase 1b study of the effects of black raspberries on rectal polyps in patients with familial adenomatous polyposis[J]. Cancer Prev Res, 2014, 7:666-674.
[37] Mensaert K, Denil S, Trooskens G, et al. Next-generation technologies and data analytical approaches for epigenomics[J]. Environmental and Molecular Mutagenesis, 2014, 55(3):155-170.
[38] Li N, Ye M, Li Y, et al. Whole genome DNA methylation analysis based on high throughput sequencing technology[J]. Methods,2010, 52(3):203-212.
[39] Rohde C, Zhang Y, Jurkowski TP, et al. Bisulfite sequencing data presentation and compilation(BDPC)web server—a useful tool for DNA methylation analysis[J]. Nucleic Acids Research, 2008,36(5):e34.
[40] Xi Y, Li W. BSMAP:whole genome bisulfite sequence MAPping program[J]. BMC Bioinformatics, 2009, 10(1):232.
[41] Down TA, Rakyan VK, Turner DJ, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis[J]. Nature Biotechnology, 2008, 26(7):779-785.
[42] Wang Y, Leung FC. An evaluation of new criteria for CpG islands in the human genome as gene markers[J]. Bioinformatics, 2004,20(7):1170-1177.
[43]Takai D, Jones PA. The CpG island searcher:a new WWW resource[J]. In Silico Biology, 2003, 3(3):235-240.
[44]Ioshikhes IP, Zhang MQ. Large-scale human promoter mapping using CpG islands[J]. Nature Genetics, 2000, 26(1):61-63.
[45]Grunau C, Renault E, Rosenthal A, et al. MethDB-a public database for DNA methylation data[J]. Nucleic Acids Research, 2001, 29(1):270-274.
[46]Grunau C, Renault E, Roizes G. DNA methylation database“MethDB”:a user guide[J]. J Nutr, 2002, 132:2435S-2439S.
[47]Amoreira C, Hindermann W, Grunau C. An improved version of the DNA Methylation database(MethDB)[J]. Nucleic Acids Research, 2003, 31(1):75-77.
[48] Negre V, Grunau C. The MethDB DAS server:adding an epigenetic information layer to the human genome[J]. Epigenetics, 2006, 1(2):101-105.
[49]Kumaki Y, Oda M, Okano M. QUMA:quantification tool for methylation analysis[J]. Nucleic Acids Research, 2008, 36(suppl 2):W170-W175.
[50] Pattyn F, Hoebeeck J, Robbrecht P, et al. methBLAST and meth-PrimerDB:web-tools for PCR based methylation analysis[J]. BMC Bioinformatics, 2006, 7(1):496.
[51]Li LC, Dahiya R. MethPrimer:designing primers for methylation PCRs[J]. Bioinformatics, 2002, 18(11):1427-1431.
[52]Laird PW. Principles and challenges of genome-wide DNA methylation analysis[J]. Nature Reviews Genetics, 2010, 11(3):191-203.
(責任編輯 狄艷紅)
Detection Methods of Genome-wide DNA Methylation Level Based on High-throughput Sequencing Technology
Ma Langlang1,2Liang Zhenjuan2Xian Xin2Luo Shiwen2Li Qingchao2Liang Qianyun2
(1. Maize Research Institute of Sichuan Agricultural University,Chengdu 611130;2. Bijie Institude of Agricultural Science,Bijie 551700)
DNA methylation has been being the research focus of epigenetics study in the recent years. There are over 10 kinds of methods for detecting DNA methylation level, and detection of methylation status on genome-wide level has been achieved with the development of the NGS(Next-Generation Sequencing). At present the major detection methods of DNA methylation level on genome-wide scope based on NGS include BSP-seq(Bisulfite Sequencing PCR), MeDIP-seq(Methylated DNA Immunoprecipitation Sequencing)and MBD-seq(Methylated DNA Binding Domain Sequencing). In this paper, the research progress of principles, procedures, advantages and disadvantages, optimization of the usage and some bioinformatics resources on these 3 methods were reviewed, which aims to provide some beneficial ideas to researchers who are studying DNA methylation models while employing methods of high-throughput sequencing.
high-throughput sequencing technology;genome-wide;DNA methylation level;BSP-seq;MeDIP-seq;MBD-seq
10.13560/j.cnki.biotech.bull.1985.2015.07.007
2014-10-19
貴州省科學技術基金計劃重點項目[黔科合JZ字(2014)2001號],州省科學技術基金計劃項目[黔科合J字(2013)2002號],貴州省畢節市農業攻關項目(畢科合19號)
馬浪浪,男,碩士,研究方向:表觀遺傳學;E-mail:sxyljxml@163.com
李清超,男,助理研究員,研究方向:玉米分子育種;E-mail:569733223@qq.com梁黔云,男,研究員,研究方向:玉米育種;E-mail:blqy678@163.com
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
人大建設(2019年12期)2019-05-21 02:55:32
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
中國火炬(2010年8期)2010-07-25 11:34:30
