二價鑭系均配金屬化合物“Open-Metallocenes”
——雙(2,4-二叔丁基戊二烯基)釤和鐿配合物
2015-12-05 07:28:15陳潤海董云會李月云
無機化學學報 2015年6期
陳潤海 王 平 劉 青 董云會 李月云
(山東理工大學化學工程學院,淄博 255049)
二價鑭系均配金屬化合物“Open-Metallocenes”
——雙(2,4-二叔丁基戊二烯基)釤和鐿配合物
陳潤海王平劉青董云會李月云*
(山東理工大學化學工程學院,淄博255049)
雙(2,4-二叔丁基戊二烯基)鑭系金屬化合物(η5-Pdl′)2Yb(THF)(1)和(η5-Pdl′)2Sm(DME)(2)可以經LnI2和2,4-二叔丁基戊二烯基鉀(K(Pdl′))由置換反應制得。K(Pdl′)和LnCl3發生氧化還原反應也能夠得到化合物1和2。對2個化合物進行了表征。X射線單晶衍射結果顯示,2個化合物中Pdl′基都是以η5模式配位到金屬上。2個化合物都能夠在室溫下引發己內酯聚合并表現出高活性,得到分子量分布窄的聚己內酯。
釤;鐿;戊二烯基;聚合;己內酯
0 Introduction
In last decades,Pdl(pentadienyl)ligand arouse lots of interest and various kinds of Pdl have been synthesizedandusedinorganometallics(“openmetallocenes”).Besides bonding to metal in a variety of η1,η3and η5modes,it was found that the usual η5-Pdl is a more sterically demanding[1],and even more strongly bonding to center metalthancyclopentadienyl[2]. Another significant feature of Pdl ligands is its great preference on bonding to metals in low oxidation especially divalence[3].The steric and electronic properties of Pdl ligands can be easily adjusted by using different substituted derivatives such as 2,4-C7H11(2,4-Me2-Pdl),1,5-(Me3Si)2-C5H5[1,4],edge-bridged pentadienyl[5],chiral pentadienyl[6],“open indenyl”[7],bridged bis(pentadienyl)[8],and even hetero-pentadienyl[9].Although the high reactivity of the electronically pentadienyl ligands has led to a wide applications in organometallic synthesis and coupling reactions[10],it is surprising that the usual bis(η5-Pdl)Smcompound,i.e. pure“open-samarocenes”,is still elusive.The classical bis-coordination mode of the Pdl ligand seems fade it′s favor when interaction with Sm.The reaction of SmCl3with K(2,4-C7H11)(Molar ratio:1∶1 or 1∶2)in THF all gave the Sm(2,4-C7H11)3compound instead of expected Sm(2,4-C7H11)Cl2or Sm(2,4-C7H11)2Cl(Scheme 1)[11].In contrast to Sm complex,the same reaction of YbCl3with K(2,4-C7H11)(Molar ratio:1∶3)led to the divalent Yb(2,4-C7H11)2(DME)(DME=dimethoxyethane) compound due to the reduction pathway[11b].This could be explained by the higher reduction potential of Sm3+/Sm2+(-1.55 V)than Yb3+/Yb2+(-1.15 V)[12].
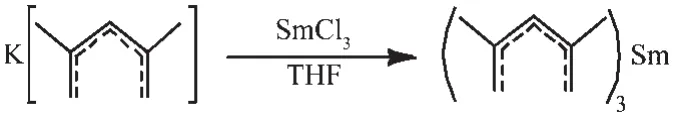
Scheme 1
There were limited examples on the pentadienyl rare earth complexes used for coordination polymerization.A systematical comparison of the stereoselectivity of these tris-,bis-and mono-(2,4-C7H11)ligated Ndcomplexesonbutadienepolymerizationhas already been reported by Geitner[13].Nevertheless,the“open-metallocenes”has not been applied in ester polymerization.In contrast to the well-known,aromaticsoluble metallocene(C5Me5)2Sm(THF)2,which has been widely used in the polymerizations of olefins[14]and esters[15],the classical pentadienyl 2,4-C7H11ligated lanthanidecomplexesshowedpoorsolubilityin common solvents,precluding the further utilization in stoichiometric or catalytic reactions[16].Introduction of steric bulkier substituents such astBu or SiMe3groups on pentadienyl ligand could enhance the solubility and stabilization of the corresponding metal complexes[17]. Herein,we report the synthesis of the novel bis(2,4-tBu2-C5H5)Smand Ybcomplexes by the salt metathesis reactions between 2,4-di-butyl-pentadienyl potassium(K(Pdl′))and lanthanide halides,and the application in ring-opening polymerization of ε-caprol-a cton.
1 Experimental
1.1General
The K(Pdl′),YbCl3,SmCl3,YbI2(THF)2and SmI2(THF)2were prepared according to the literature[17b,18]. All solvents were purified with an MBraun SPS system.Organometallic samples for NMR spectroscopic measurements were prepared in the glovebox by use of NMR tubes sealed by paraffin film.NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer.All the subsequent operations were carried out in the gloves box or Schleck line under N2atmosphere.
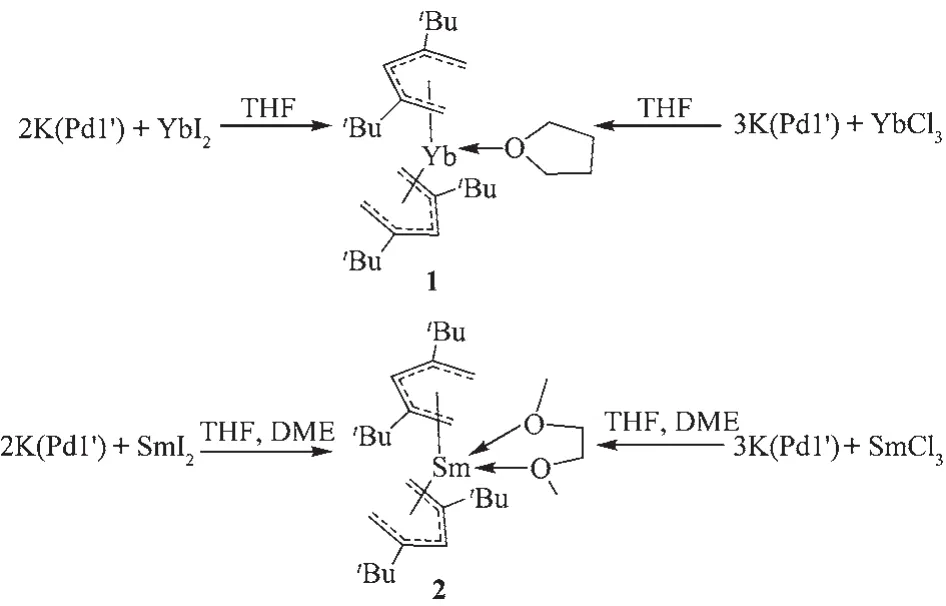
Scheme 2 Syntheses of complexes 1 and 2
1.2Synthesis of(2,4-(tBu)2-C5H5)2Yb(THF)(1) Path A:From YbI2(THF)2and K(Pdl′)
K(Pdl′)(0.38 g,1.76 mmol)was added to a slurry of YbI2(THF)2(0.50 g,0.88 mmol)in 20 mL THF.The solution turned dark green immediately,and the suspension was stirred for 1 h at room temperature. All the volatiles were removed in vacuo.The residue was extracted with three portions of hexane(5 mL). The extraction was filtered,concentrated to ca.5 mL and then kept under-30℃.Malachite green crystals of complex 1 were collected after several days.Yield: 0.385 g(69%).Anal.Calcd.for C30H54OYb(%):C, 59.68;H,9.01;O,2.65.Found(%):C,59.36;H,9.13; O,2.76.1H NMR(400 MHz,C6D6,298 K):δ 4.67(t, 2H,CH,4JH-H=2.2 Hz),4.32(d,4H,CH2,4JH-H=2.3 Hz),3.45(br,8H,CH2,α-H in THF),1.33(br,40H, (CH3)3C,β-H in THF).
Path B:From YbCl3and K(Pdl′)
To a slurry of YbCl3(0.14 g,0.5 mmol)in 20 mL THF was added K(Pdl′)(0.33 g,1.5 mmol),the stirring solution rapidly turned dark green,and the suspension was stirred for 2 h at room temperature. After removal of all the volatiles,the residue was extracted with three portions of hexane(5 mL).The extraction was filtered,concentrated to ca.3 mL,and kept at-30℃overnight to give the malachite green crystals.Yield:0.08 g(26%).1H NMR data is consist with complex 1.
1.3Synthesis of(2,4-(tBu)2-C5H5)2Sm(DME)(2)
Path A:From SmI2(THF)2and K(Pdl′)
To a slurry of SmI2(THF)2(1.25 g,2.29 mmol)in 50 mL THF was added K(Pdl′)(1 g,4.58 mmol),the solution turned black immediately,and the suspension was stirred for 2 h at room temperature.All the volatiles were removed in vacuo.The residue was extracted with three portions of hexane(10 mL).The extraction was filtered,concentrated to ca.8 mL. Addition of 0.5 mL DME to the solution afforded a large amount of black precipitate immediately.After removal of supernatant solution,the left black solid was washed with hexane,and dried under vaccum to give complex 2.Yield:1.0 g(80%).Single crystals of complex 2 suitable for X-ray analysis were grown from DME solution at-30℃.Anal.Calcd.for C30H56O2Sm (%):C,60.14;H,9.42;O,5.34.Found(%):C,60.50; H,9.33;O,5.21.
Path B:From SmCl3and K(Pdl′)
To a slurry of SmCl3(0.13 g,0.5 mmol)in 20 mL THF was added 3 equivalent of K(Pdl′)(0.33 g,1.5 mmol),the stirring solution rapidly turned black,and the suspension was stirred for 2 h at room temperature. After removal of all the volatiles,the residue was extracted with three portions of hexane(5 mL).The extraction was filtered,concentrated to ca.3 mL. Addition of 0.5 mL DME to the solution afforded a large amount of black solid.Recrystallization from DME at-30℃overnight gave the black brick crystals.Yield:0.08 g(27%).X-Ray data analysis disclosed that the cell parameters are the same with complex 2.
1.4Ring-opening polymerization of ε-caprolactone
All polymerization reactions were carried out in the glovebox under N2atmosphere.In a typical procedure,ε-caprolactone was added to the toluene solution of the initiator 1 and 2 with vigorous magnetic stirring at the desired temperature.After a certain time,the polymerization was quenched with acidified methanol, and the polymer wasprecipitated.The resulting polymer was washed with methanol and dried in a vacuum at 35℃.
1.5X-ray crystallographic studies
Crystals for X-ray analysis were obtained as described in the preparations.The crystals were manipulated in a glovebox.Data collections were performed at-88.5℃on a Bruker SMART APEX diffractometer with aCCD area detector,using graphite-monochromatic Mo Kα radiation(λ=0.071 073 nm).The determination of crystal class and unit cell parameters was carried out by the SMART program package.The raw frame data were processed using SAINT and SADABS to yield the reflection data file[19].The structures were solved by using the SHELXTL program[20].Refinement was performed on F2anisotropically for all non-hydrogen atoms by the full-matrix least-squares method. The hydrogen atoms were placed at the calculated positions and were included in the structure calculation without further refinement of the parameters.The methyl carbon atoms(C7,C8,C9 and C11,C12,C13) of tert-butyl groups and the carbon atoms(C27,C28, C29,C30)of THF fragment were disordered and were refined with 64%,52%and 50%occupancy anisotropically respectively for all the atoms in complex 2.
CCDC:1045037,1;1045038,2.
2 Results and discussion
2.1Syntheses and crystal structures
As our expectation,the bulkier 2,4-tBu2-Pdl could more efficiently stabilize Lnthan the 2,4-Me2-Pdl. Salt metathesis reactions of LnI2(Ln=Sm,Yb)with two equivalents of K(Pdl′)proceeded smoothly in THF to afford corresponding“open-metallocenes”(Pdl′)2Sm (DME)and(Pdl′)2Yb(THF)in moderateyields,respectively.Noteworthily,these two complexes showed higher solubility in hydrocarbon than the previous reported complex Yb(2,4-C7H11)2(DME),which was insolublein hexane[11b].The introduction of DME afforded an improved stability of the Smcomplex. After the addition of DME,the solubility of 2 in hexane decreased dramatically,and great amount of 2 precipitated from the solution.It could be assumed that the bi-coordinate sites of DME molecular stabilized the entire complex,and decreased the solubility of the coordinated Sm complex.The reactions of K(Pdl′) with SmCl3or YbCl3in a molar ratio of 3∶1,also afforded a blackish and a dark green solution,suggested that reductive ways rather than salt elimination reactions happened.ThesubsequentX-raycrystallographic studies of these single crystals product confirm the reductive path ways.This result is different from the previous reaction between the K(C7H11)and SmCl3, which afforded the Sm(C7H11)3complex.It could be inferred that sterically demandingtBu enhanced the stabilization of the divalent samarium center.
Due to the striking similarity in the chemistry of Yband Cacomplexes,in the1H NMR spectrum of the complex 1,the chemical shifts of Pdl fragments are almost the same as(Pdl′)2Ca(THF),which suggested a η5-U coordination of the 2,4-di-butyl-pentadienyl ligand[21].However,the chemical shifts of coordinated THF move slightly low-field.The resonance of α-H and β-H of THF fragment occasionally overlaps with the prime carbon H(exo)resonance(δ 3.45)and thetBu protons(δ 1.33)of Pdl′ligands,respectively. Unfortunately,complex 2 could not be characterized by1H NMR spectra because of its paramagnetic property.
The molecular structures of 1 and 2 are shown in Fig.1,and the relevant bond distances and angles are listed in Table1.In the pentadienyl fragments,the CCdistancesshowadistinctshort-long-long-short pattern.The carbon atoms lie in a plane,adopting a η5coordination mode.The Yb-C distances of complex 1 are the same as the previous analog(2,4-Me2-C5H5)2Yb(DME).The conformation angle χ of these two complexes,defined as the angle between the two planes(centroid(C1-C5)-C3-M)and(centroid(C14-C18)-C16-M),are all approximately 180°.This antieclipsed conformation,which displays the mutual orientations of the two pentadienyl moieties,are consistent with Ca,Cr and Fe pentadiene analog[17b], but in contrast with(2,4-Me2-C5H5)2Yb(DME).The interactionofthebulkiertBudeterminesthe orientation of the pentadienyl to offer a rather flat potentialenergysurfacewithrespecttothe conformation angle χ.
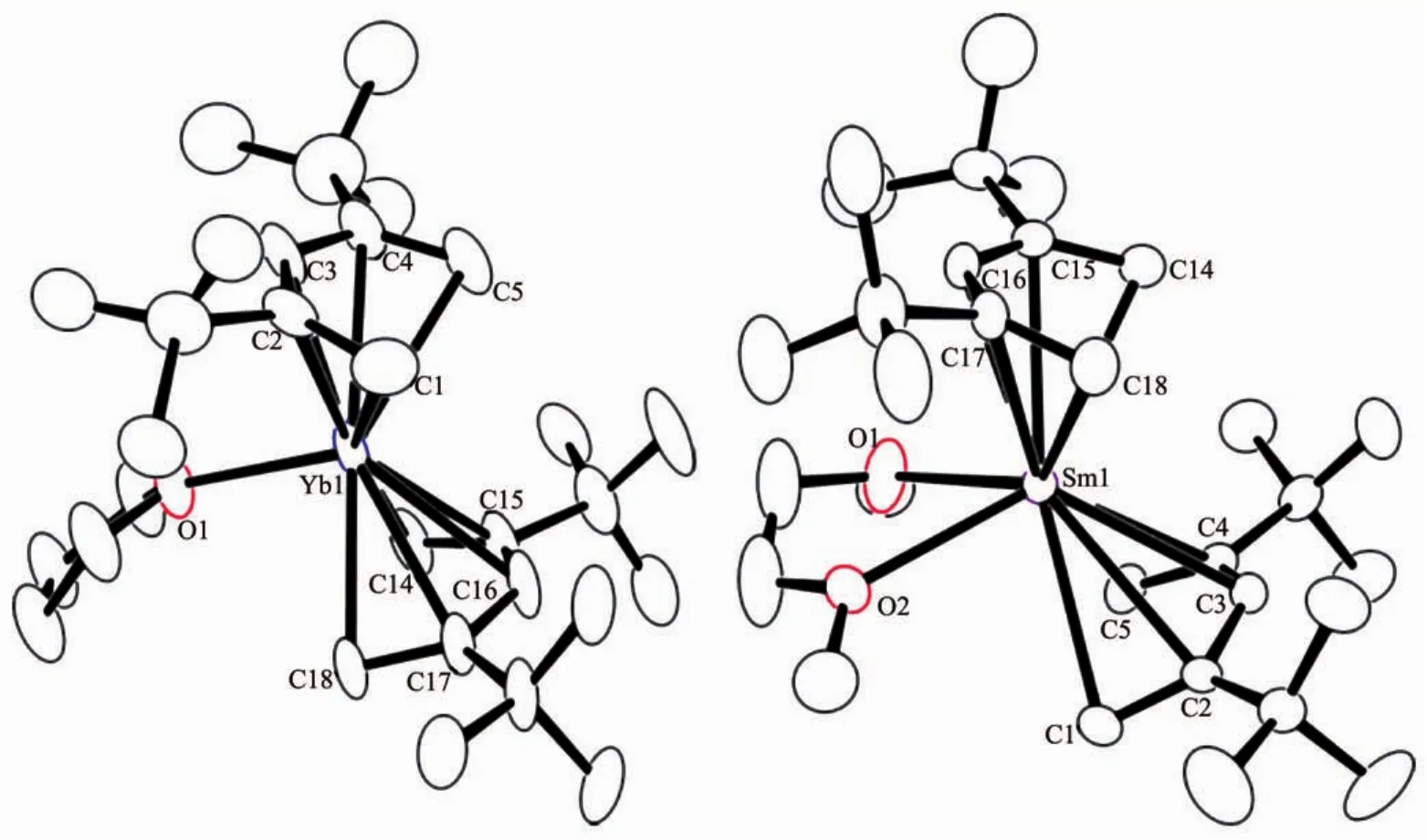
Fig.1 ORTEP diagrams of 1 and 2(10%and 35%probability ellipsoids respectively)
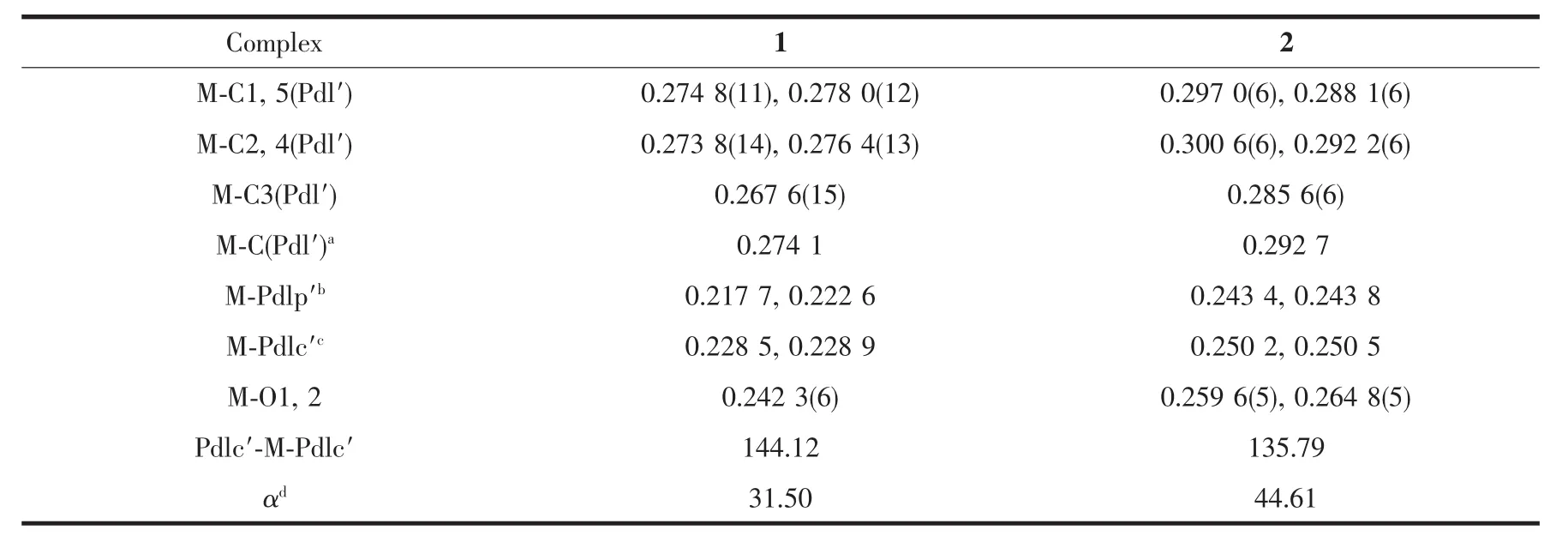
Table1 Selected bond distances(nm)and angles(°)of 1 and 2
2.2Polymerization of ε-caprolactone
Ring-opening polymerization ofε-caprolactone (ε-CL)under varying conditions was investigated by using complexes 1 and 2.The representative polymerization results are summarized in Table2.Complexes 1 and 2 both display high activity for the ring-opening polymerization of ε-CL in toluene at room temperature. As the monomer was introduced,the dark solution immediately faded to colorless due to an oxidation process,indicating the transformation of Ln2+to Ln3+. Complexes 1 and 2 showed much higher activity toward ε-CL polymerization than that of(C5Me5)2Sm (THF)2[15].The monomer conversion could be achieved in 1 min rather than 1 h for(C5Me5)2Sm(THF)2in the same condition.The molecular weights of the obtained polymers increase from 7 700 to 26 200 with monomer -to-initiator ratios increasing from 100 to 400 with a moderate PDI(1.24~1.62).One of the factors to explain such disparity is that these two seemingly similarligandsadopteddifferentπ-delocalization modes.Once the caprolactone introduced,a η5-η3-η1allyl transformation of the pentadienyl could take place more readily than cyclopetadienyl due to the small loss of resonance delocalization energy.These results are compared to the previous investigation of ethylenepolymerizationcatalyzedbypentadienyl chromium complexes.

Table2 Polymerization of ε-caprolactone by complexes 1 and 2a
3 Conclusions
Hydrocarbon soluable bis(2,4-tBu2-pentadienyl) Sm and Yb complexes have been synthesized and crystallographically characterized.Crystal structures disclose the η5-pentadienyl-metal bonding mode.The introduction oftBu in the 2,4-position of pentadiene enhancedthestericallydemanding,provideda promise for stabilized the divalent Yb and Sm centre. Comparedwithtypicalsamariummetallocene, complexes 1 and 2 showed higher activities toward εcaprolactone polymerization with narrow polydispersities.
Supporting information(The crystal data and structure refinement for complexes 1 and 2,1H NMR of complexes 1)is available at http://www.wjhxxb.cn
Acknowledgements:This study was supported by the National Natural Science Foundation of China(No.21405095) and the Project of Shandong Province Higher Educational Science and Technology Program(No.J14LC09).All of the authors express their deep thanks.
[1]Ernst R D.Chem.Rev.,1988,88:1255-1291
[2]Melendez E,Arif A M,Ernst R D,et al.Angew.Chem.Int. Ed.,1988,27:1099-1101
[3]Ernst R D.Comments Inorg.Chem.,1999,21:285-325
[4](a)Ernst R D.Struct.Bond.,1984,57:1-53 (b)Ernst R D.Acc.Chem.Res.,1985,18:56-62
[5]Wilson A M,Ernst R D,et al.J.Am.Chem.Soc.,1995,117: 8490-8491
[6]Fecker A C,Craciun B F,et al.Organometallics,2014,33: 3792-3803
[7]Gloeckner A,Bannenberg,et al.Organometallics,2012,31: 4480-4494
[8]Weng W Q,Kunze K,Ernst R D,et al.Organometallics,1991, 10:3643-3647
[9](a)Reyna-Madrigal A,Ernst R D,et al.Organometallics,2012, 31:7125-7145 (b)Trakarnpruk W,Arif A M,Ernst R D.Organometallics, 1992,11:1686-1692
[10]Rajapakshe A,Gruhn N E,Lichtenberger D L,et al.J.Am. Chem.Soc.,2004,126:14105-14116
[11](a)Jin J,Jin S,Chen W,et al.J.Chem.Soc.Chem.Commun., 1991:1328-1329 (b)Baudry D,Nief F,Ricard L.J.Organomet.Chem.,1994, 482:125-130
[12]Nief F.Dalton Trans.,2010,39:6589-6598
[13](a)Sieler J,Simon A,Peters K,et al.J.Organomet.Chem., 1989,362:297-303 (b)Kunze M R,Steinborn D,Merzweiler K,et al.Z.Anorg. Allg.Chem.,2007,633:1451-1463
[14]Kaita S,Hou Z M,Wakatsuki Y.Macromolecules,1999,32: 9078-9079
[15]Evans W J,Katsumata H.Macromolecules,1994,27:2330-2332
[16]Overby J S,Hanusa T P.Angew.Chem.Int.Ed.,1994,33: 2191-2193
[17](a)Gedridge R W,Arif A M,Ernst R D.J.Organomet.Chem., 1995,501:95-100 (b)Reiners M,Fecker A C,et al.Dalton Trans.,2014,43: 6614-6617
[18](a)Girard P,Namy J L,Kagan H B.J.Am.Chem.Soc., 1980,102:2693-2698 (b)Taylor M D,Carter C P.J.Inorg.Nucl.Chem.,1962,24: 387-391
[19]Sheldrick G M.SADABS,Empirical Absorption Correction Program,Bruker Analytical X-ray Systems,Madison,WI, 1996.
[20]Sheldrick G M.SHELXTL Version6.12,Bruker Analytical X-Ray Systems,Inc.,Madison,WI,2001.
[21]Wilson D R,Dilullo A A,Ernst R D.J.Am.Chem.Soc., 1980,102:5928-5930
Homoleptic Divalent Lanthanide“Open-Metallocenes”—Bis(2,4-tBu2-pentadienyl)Sm and Yb Complexes
CHEN Run-HaiWANG PingLIU QingDONG Yun-HuiLI Yue-Yun*
(School of Chemical Engineering,Shandong University of Technology,Zibo,Shandong 255049,China)
The bis(2,4-tBu2-pentadienyl)lanthanide complexes(η5-Pdl′)2Yb(THF)(1)and(η5-Pdl′)2Sm(DME)(2) were successfully prepared in high yield by direct salt metathesis between LnI2and 2,4-di-butyl-pentadienyl potassium(K(Pdl′)).Complexes 1 and 2 could also be obtained smoothly from the reaction of LnCl3and K(Pdl′) through oxidation-reduction reaction.These two compounds were characterized and the X-ray single crystal analysis revealed a predominantly η5-pentadienyl-metal bonding.Both two compounds could initiate ring-opening polymerization of ε-caprolactone rapidly at room temperature and led to narrow polydispersities with high activity. CCDC:1045037,1;1045038,2.
Samarium;Ytterbium;pentadienyl;polymerization;ε-caprolactone
O614.33+7;O614.346
A
1001-4861(2015)06-1239-06
10.11862/CJIC.2015.169
2015-02-15。收修改稿日期:2015-04-20。
國國家自然科學基金(No.21405095),山東省高等學校科技計劃項目(No.J14LC09)資助項目。
*通訊聯系人。E-mail:liyueyun71@163.com