EMIM離子液體離子簇模型的量子化學計算
2015-12-05 06:29:49戚傳松
物理化學學報 2015年9期
李 巍 張 靜 戚傳松
(1北京石油化工學院化學工程學院, 北京 102617; 2中國科學院大學材料科學與光電技術學院, 北京 100049)
EMIM離子液體離子簇模型的量子化學計算
李 巍1,*張 靜2戚傳松1
(1北京石油化工學院化學工程學院, 北京 102617;2中國科學院大學材料科學與光電技術學院, 北京 100049)
以1-乙基-3-甲基咪唑(EMIM)鹵化物、氟硼酸鹽、三溴化物和二碘溴酸鹽、氯鋁酸和溴鋁酸鹽等不同種類EMIM離子液體為研究對象, 對多陽離子、多陰離子的離子簇模型進行了量子化學計算研究. 首先在B3LYP/6-311++G(d,p)水平上(I使用6-311G(d,p)基組)對{[EMIM]Xn}(n–1)–(X = Cl, Br, I, BF4, AlCl4, AlBr4, Br3, IBrI, FHF; n = 2, 3)和{[EMIM]2Xn'}(n'–2)–(n' = 3, 4, 5)離子簇進行構型優化, 并對鹵化物和氟硼酸鹽進行了振動光譜計算. 結果表明所采用理論模型在鍵長、鍵角等結構參數及紅外振動光譜方面均與實驗結果符合較好.同時對不同離子簇模型中陰、陽離子間相互作用能與實驗熔點之間的關系進行了研究, 發現采用{[EMIM]2Xn'}(n'–2)–模型時EMIM離子液體實驗熔點與陰、陽離子間相互作用能之間呈現近線性關系.
密度泛函理論; 咪唑離子液體; 相互作用能; 離子簇; 熔點
1 引 言
室溫離子液體(RTILs, 簡稱ILs)是由不對稱的有機陽離子與無機或有機陰離子構成的一類新型溶劑, 由于其環保性好等特點被認為是傳統有機溶劑的有力替代者;1–4而增加離子液體的種類、充實離子液體化學的基礎內容、開辟離子液體新的研究與應用領域是綠色化學的需要. 有機陽離子與陰離子之間可能的組合數目龐大, 但是能夠滿足特定應用需求的組合方式是有限的, 因而離子液體經常被稱為“designer solvents”, 而如何設計出具有特定性質及功能的離子液體是當前的一個研究熱點. 目前離子液體的設計仍然是以一種比較盲目的“tryand-error”模式進行, 并沒有真正做到根據目標進行設計. 為了能夠從分子水平上設計低熔點、功能化的離子液體, 以及在新型離子液體的探索中避免人力及物力的浪費, 離子液體基礎性質及功能預測的理論研究勢在必行.
而離子液體的理論計算研究相對于其實驗研究發展較慢, 這主要是由于對于凝聚相體系至今還沒有發展出一個高精度和高效性兩者兼備的算法.目前常用的理論手段包括使用量子化學計算方法研究氣相中離子對結構, 或是構建力場、利用分子模擬方法研究離子液體的宏觀物理性質, 以及利用統計方法結合量子化學5–14或半經驗方法15–19進行定量構效關系(QSPR)研究. 在過去的10年里, 基于分子描述符的QSPR研究發展迅速, 在相關的研究中多數采用半經驗方法或分子力學方法優化構型, 參數的選擇主要服務于精度的需要, 對預測結果影響較大的參數數量較多, 對物理化學性質的影響因素只能定性的分析; 同時, 方法應用的效果依賴于算法及個人經驗, 至今為止還沒有一種普適于不同種類離子液體的構效關系表達式.
在構效關系研究方面我們的目標是力圖發現一種普適性強、形式簡單、物理意義明確的構效關系式, 而目前我們最關注的實驗性質是熔點, 這是由于低熔點是離子液體具有應用前景的首要條件; 此外He等20發現對于N-取代的甘氨酸酯化物陽離子型離子液體, 熔點與黏度呈現正相關性; Rahman等21在研究四乙銨氨基酸離子液體時則發現其離子導電性與黏度成線性相關關系. 可見, 熔點是離子液體非常重要的一項性質指標. 考慮到離子液體是一種以庫侖相互作用為主的體系, 而在發生熔化過程體積發生膨脹時需要克服的阻力以陰陽離子間相互作用為主, 因此我們以量子化學計算獲得的離子間相互作用為唯一的描述符, 試圖建立離子液體的熔點與離子間相互作用的定量關系. 我們已經在氨基酸陽離子型離子液體、5烷基咪唑氟硼酸離子液體6和烷基咪唑鹵化物7中進行了嘗試并獲得一定的成功. 類似的嘗試也表現在部分其他小組的工作中, 但多數以失敗告終, 其中Wu8和Mohajeri9等均對咪唑氨基酸離子液體的離子對進行了系統地量子化學計算研究; 而Turner10及Katsyuba小組11分別系統研究了咪唑鹵化物及咪唑氟硼酸系列離子液體的離子對結構. 上述工作在咪唑離子液體研究時未能獲得與實驗熔點與某種分子結構性質的相關性的主要原因是由于他們均采用單個陽離子與單個陰離子組成的離子對模型, 而此模型不能真正反映凝聚相體系中真實的結構特征.6,7我們之前在研究咪唑類化合物時選擇一個咪唑陽離子與2–3個陰離子組成的一價或二價分子離子為研究對象, 發現量子化學計算優化獲得的結構與晶體結構實驗結果更加符合, 且陰、陽離子間相互作用能與實驗熔點之間存在線性關系.6,7在之前關于咪唑類離子液體的構效關系研究中, 基本是以單一種類陰離子的體系為研究對象. 在本文中, 我們在前期工作的基礎上擴展研究對象, 對陰離子種類不同的常見1-乙基-3-甲基咪唑(EMIM)離子液體進行量子化學計算研究, 采用多個陽離子和多個陰離子組成的離子團簇模型,考慮不同陰離子與EMIM陽離子間的作用模式, 并進一步研究熔點與離子間相互作用能之間的構效關系.
2 研究體系及研究方法
2.1 研究體系
我們之前研究了陰離子相同的咪唑類離子液體實驗熔點與離子間相互作用能之間的關系, 在本次工作中我們選擇陽離子均為1-乙基-3-甲基咪唑的離子液體為研究對象, 通過檢索Reaxys數據庫22收集具備實驗熔點的1-乙基-3-甲基咪唑化合物; 同時結合檢索劍橋晶體數據庫, 選擇既具有實驗熔點又具備晶體結構信息的常見1-乙基-3-甲基咪唑化合物作為研究對象, 具體包括: 氟硼酸鹽([EMIM][BF4])、氟化物([EMIM][FHF])、氯化物([EMIM]Cl)、溴化物([EMIM]Br)、碘化物([EMIM]I)、三溴化物([EMIM][Br3])及溴鋁酸鹽([EMIM][AlBr4]). 此外, 氯鋁酸鹽([EMIM][AlCl4])、二碘溴酸鹽([EMIM][IBrI])雖然未查到晶體結構信息, 但由于其陰離子部分與已知晶體結構的化合物具有相似性, 在本文工作中也進行了討論.
2.2 理論模型的選擇
通過檢索劍橋晶體數據庫, 得到了百余種1-乙基-3-甲基咪唑化合物的晶體結構信息. 通過觀察、對比發現陰離子的尺寸、對稱性對晶體中離子的排列有著至關重要的影響, 從而進一步影響離子間的相互作用模式. 本文討論的常見EMIM離子液體中陰離子從小至大排列為(括號內數值為估算得到的離子外接球半徑, 其中多原子陰離子按照晶體結構中鍵長數據及端原子范德華半徑23估算得到): Cl–(0.18 nm)、Br–(0.20 nm)、I–(0.22 nm)、FHF–(0.25 nm)、(0.44 nm). 陰離子尺寸及對稱性相近時晶體結構中陰、陽離子的排布模式也相似. 我們選擇能夠反映晶體結構中陰、陽離子相互作用模式的最小結構單元作為理論模型, 具體選擇的離子簇如圖1和圖2所示,具體分析詳見結果與討論部分.
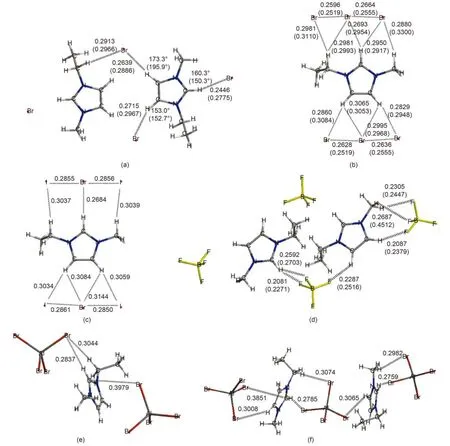
圖1 EMIM離子液體離子簇優化構型Fig.1 Optimized structures of ion clusters of EMIM ILs
在我們之前的工作6,7中曾經對烷基咪唑鹵化物及烷基咪唑氟硼酸鹽進行過系統的量子化學計算研究, 當時采用的模型是{[EMIM]Xn}(n–1)–(n=2, 3),這一模型比較好地反映了直接與咪唑陽離子作用的周圍陰離子的排布情況, 并且從該模型出發得到了離子間相互作用能與實驗熔點之間的線性關系.但該模型仍然存在不足: 首先, 它并不適用于較大陰離子體系, 如[EMIM][Br3]、[EMIM][AlBr4]等情況; 此外, 對于氟硼酸體系該模型中B原子與咪唑環幾乎在同一平面內, 與實際晶體結構不完全一致.在本文中我們將選取能夠更好反映實際晶體結構特征的離子簇模型作為研究對象, 探究陰離子種類不同的咪唑離子液體中離子間的作用模式.
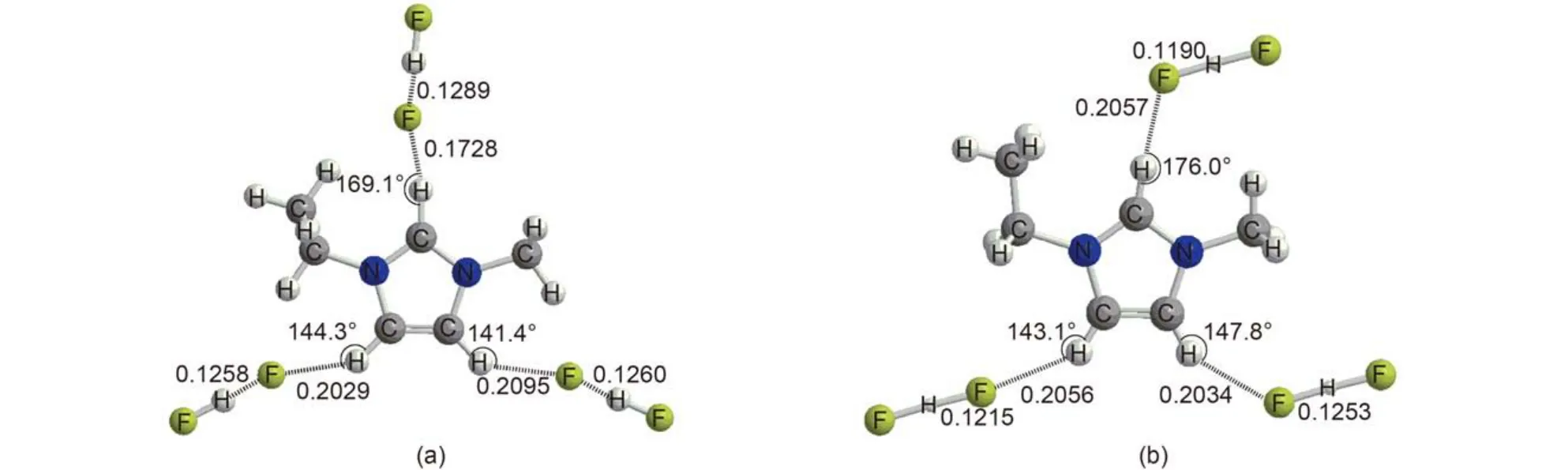
圖2 {[EMIM][FHF]3}2–優化構型及晶體結構中相應結構單元Fig.2 Optimized geometry of {[EMIM][FHF]3}2–and corresponding geometry in crystal
2.3 量子化學計算
基于Gaussian 03軟件包,24采用密度泛函理論(DFT)方法B3LYP和6-311++G(d,p)基組(I使用6-311G(d,p)基組), 對不同離子簇進行了構型優化并對部分離子簇優化構型進行了振動頻率分析, 將得到的理論振動光譜與實驗結果進行對比, 幫助指認實驗紅外光譜譜線的同時對不同理論模型的結果進行了對比. 此外, 利用超分子方法計算了離子簇中離子間相互作用能, 即分別代表經過結構優化的陽離子、陰離子及離子簇能量),25并且采用Counterpoise方法進行了基組重疊效應(BSSE)修正,26–28進而討論離子間相互作用能與實驗熔點之間的關系.
3 結果與討論
3.1 優化構型及振動光譜
各離子簇的優化構型見圖1及圖2. 其中部分重要結構參數列于圖中(圖中括號部分為實驗晶體結構中對應的數值, 鍵長量綱為nm)及相應表格中. 下面將對不同的體系進行具體分析.
3.1.1 鹵化物
{[EMIM]2Br4}2–的優化構型參見圖1(a), 碘化物與氯化物的優化構型和溴化物相似, 未給出具體構型圖, 相應結構參數列于表1中(優化結果具有對稱性, 僅列出一組數據). 如圖1(a)所示的結構單元采用兩種不同的取向在空間交錯排布構成周期性結構.
溴化物與碘化物的優化結果均與晶體結構中的相應結構參數符合較好, 從圖1(a)及表1列出的具體數值分析, Br…H距離理論結果比實驗值小0.025–0.033 nm, Br…C距離均比實驗值小0.010 nm; I…C的距離與實驗值比較小0.005 nm左右, 這主要是由于未考慮周圍離子環境而孤立考慮離子簇所造成的. 之前氯化物晶體結構的報道中C…Cl的平均鍵長為0.355 nm,29優化結構中C…Cl的平均值為0.346 nm, 與實驗值符合較好.
為了進行對比, {[EMIM]X3}2–(X = Br, I)模型的優化構型參數7也列于表1中. 可以發現{[EMIM]2X4}2–和{[EMIM]X3}2–模型中的結構參數都比較好地接近實驗晶體結構中的數據, 而{[EMIM]2X4}2–模型在鍵角描述方面更接近實驗值, 且從描述離子間的相互作用模式來說更全面, 同時從后面3.2節的討論中還可以看到采用{[EMIM]2X4}2–模型后其離子間相互作用能與實驗熔點之間的定量關系與其他種類陰離子的EMIM化合物符合更好.
鹵化物振動光譜的實驗數據及不同離子簇模型的理論計算值列于表2中, 其中對于實驗光譜峰的指認主要是根據與{[EMIM]2X4}2–模型理論值的比較分析來實現. 各離子簇模型的計算結果均無虛頻. 由于{[EMIM]2X4}2–模型相對于其在晶體中的結構更加緊湊, 可以看到咪唑環C-H的伸縮振動頻率雖然接近實驗值但略高于實驗值. 表2中涉及甲基和乙基的振動模式的理論計算結果明顯偏離實驗結果, 這主要是因為所采用模型并未考慮甲基和乙基周圍環境的影響. 我們根據Scott和Radom30的處理方法, 利用{[EMIM]2X4}2–(X = Cl, Br, I)模型的甲基和乙基振動模式的計算結果, 得到校正因子0.9384.表2中括號數值即為原有計算結果乘以校正因子后的數值, 經校正后與實驗結果符合較好. 而其他振動模式, 如咪唑環上的C-H伸縮振動等, 是在考慮了周圍離子環境條件下計算得到的, 因此未作進一步校正. [EMIM]Cl離子對的最穩構型及{[EMIM]Cl3}2–模型的振動光譜計算結果也列于表2中, 其振動模式與{[EMIM]2X4}2–模型不同的直接列于相應數值下方. 通過對比不難發現各模型在咪唑環甲基和乙基的振動情況描述方面差別不大; 但在咪唑環C―H的伸縮振動描述上本研究中所采用的模型與實驗值31符合更好, 而離子對模型的結果明顯偏離實驗值.
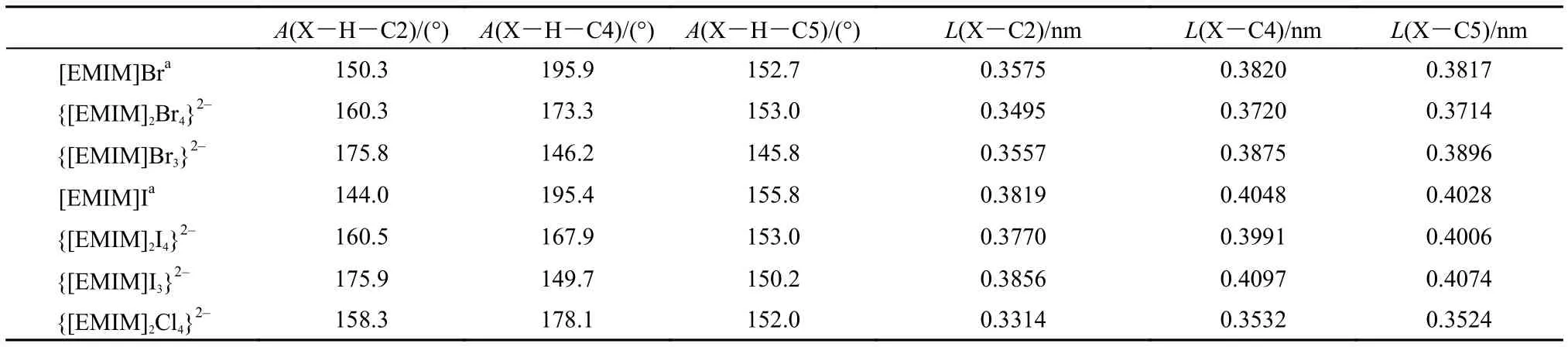
表1 EMIM鹵化物晶體結構及各優化構型結構參數Table1 Structural parameters in crystal and optimized ion clusters of EMIM halides
3.1.2 氟硼酸鹽
與鹵化物情況相似, 在EMIM氟硼酸鹽晶體結構中, 如圖1(d)所示的結構單元采用兩種不同取向在空間交錯排布形成周期性結構. {[EMIM]2[BF4]4}2–優化構型與實驗晶體結構中的相應單元比較, 咪唑環的排布相對松散: 晶體結構中咪唑環中心連線與咪唑環夾角大于{[EMIM]2[BF4]4}2–優化構型中的相應夾角(均取銳角); 咪唑環之間垂直距離實驗值比理論模型值短約0.01 nm. 在我們之前工作中采用的{[EMIM][BF4]3}2–模型中陰離子的中心B原子與咪唑環幾乎在同一平面, 而{[EMIM]2[BF4]4}2–優化構型中與咪唑環C2原子相近的陰離子中心B原子與EMIM咪唑環不同面, 后者更加符合晶體結構中的離子排布特征.
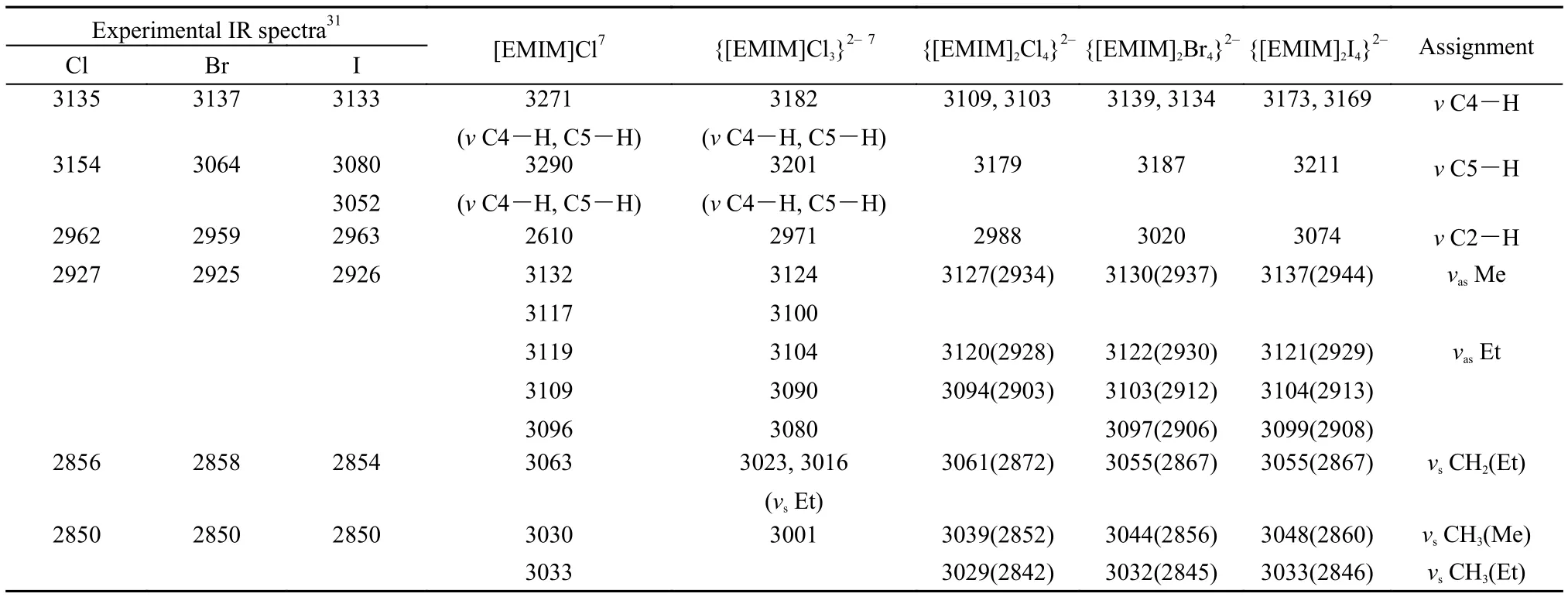
表2 [EMIM]X紅外光譜實驗及理論計算值(單位cm–1)Table2 Experimental and calculated IR spectrum data (unit in cm–1) of [EMIM]X
對EMIM氟硼酸鹽的簇模型也進行了振動頻率計算, 雖然{[EMIM]2[BF4]4}2–的計算結果中存在虛頻, 但考慮到該模型的初始構型是從EMIM氟硼酸晶體結構中的相應單元出發, 因此沒有考慮其他可能的構型. EMIM氟硼酸鹽的紅外光譜實驗值11及不同理論模型計算值列于表3中. Katsyuba等11對[EMIM][BF4]離子對模型進行了細致的研究, 但無論哪種離子對的穩定構型其紅外光譜中缺失對應于實驗值1619 cm–1的振動模式, 在我們的離子簇模型中指認該振動模式為咪唑環C2-N3與C4-C5的反對稱協同振動; 此外, 該文獻中1576 cm–1被指認為C=C振動, 而我們的計算結果中其對應于N1-C2-N3反對稱伸縮. 由于所研究離子簇模型未考慮周圍其他離子的影響, 得到的陰、陽離子間距離小于實驗值, 咪唑環C-H振動頻率均高于實驗值. 咪唑甲基和乙基的相應振動頻率采用鹵化物中得到的修正因子0.9384進行修正(表3中括號中數值),修正后結果與實驗值符合較好.
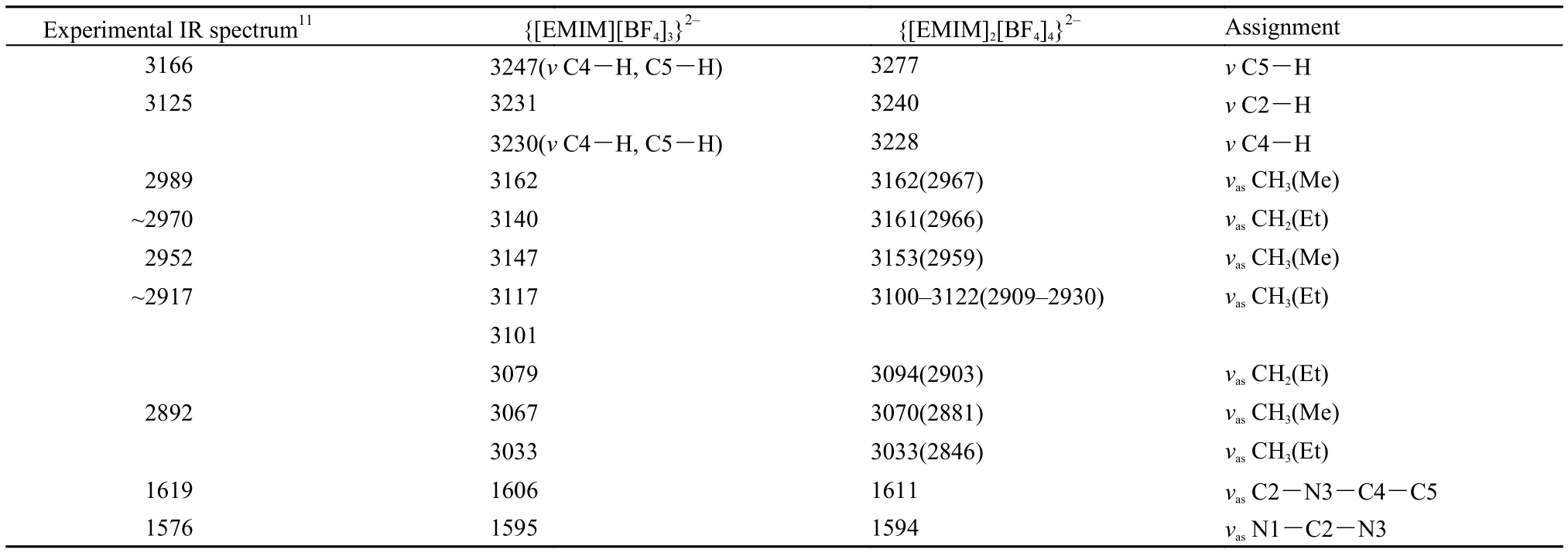
表3 [EMIM][BF4]紅外光譜實驗及理論計算值Table3 Experimental and calculated IR spectrum data of [EMIM][BF4]
3.1.3 氟化物
對于線性陰離子, 如FHF–和離子, 其與EMIM形成的化合物晶體結構呈層狀排布; 但由于二者尺寸相差較大, 每層中離子的排布方式并不相同. 在[EMIM][FHF]的晶體結構中, 每一層中每個陽離子直接與3個陰離子相互作用, 而每個陰離子直接與3個陽離子直接發生相互作用. {[EMIM][FHF]3}2–的優化構型和晶體結構中的重復單元結構如圖2所示. 從圖中可以看出由于空間排布的需要, 位于C2及C4附近的陰離子取向相對于氣相中{[EMIM][FHF]3}2–單獨存在時發生改變.
3.1.4 三溴化物及二碘溴酸鹽
[EMIM][Br3]晶體結構中沿c軸方向咪唑陽離子與交錯呈列狀排布, 根據這一特點選擇如圖1(b)所示的{[EMIM][Br3]2}–離子簇模型并進行優化.由圖中數據可見優化構型與相應晶體結構中對應部分的差別主要在于離子簇中咪唑環上方略偏向乙基一側, 下方略偏離直線結構(鍵角為173.8°), 同時Br3–在所研究離子簇中更加松散. 類似地對{[EMIM][IBrI]2}–進行結構優化, 結果列于圖1(c). 同時為了建立離子間相互作用能與實驗熔點之間的定量關系, 對呈列狀排布的{[EMIM]2[Br3]3}–和{[EMIM]2[IBrI]3}–模型也進行了優化和能量計算.
3.1.5 氯鋁酸鹽及溴鋁酸鹽
對于EMIM溴鋁酸鹽, 陰、陽離子尺寸相當, 晶體結構中離子的空間排布更加規律, 陰、陽離子沿a軸及c軸均呈列狀排布, 且陰、陽離子列呈鋸齒狀交替排布(參見圖1(f)). 本文中研究的離子簇模型描述陰、陽離子交替排布的情況.
{[EMIM][AlBr4]2}–優化構型見圖1(e), 具體結構參數列于表4(對陰離子部分相同原子進行編號時按照從左至右自小而大的方式), 優化結果基本能夠反映出晶體結構中離子間的作用模式. 相對于前面的離子簇模型, 此模型的優化構型參數偏離實驗數據較大, 最明顯的偏離是兩個陰離子的中心Al所成直線與咪唑環平面之間的夾角(銳角)比晶體結構中的相應數值小, 因而表4中二面角及鍵角數據與實驗值偏離較大, 相應的鍵長偏離實驗數據~0.07 nm. 當陰離子尺寸變大, 離子堆疊情況更加復雜時, 周圍離子的影響越來越不可忽略. 同時對{[EMIM]2[AlX4]3}–(X = Cl, Br)進行了構型優化, {[EMIM]2[AlBr4]3}–的優化結果參見圖1(f).
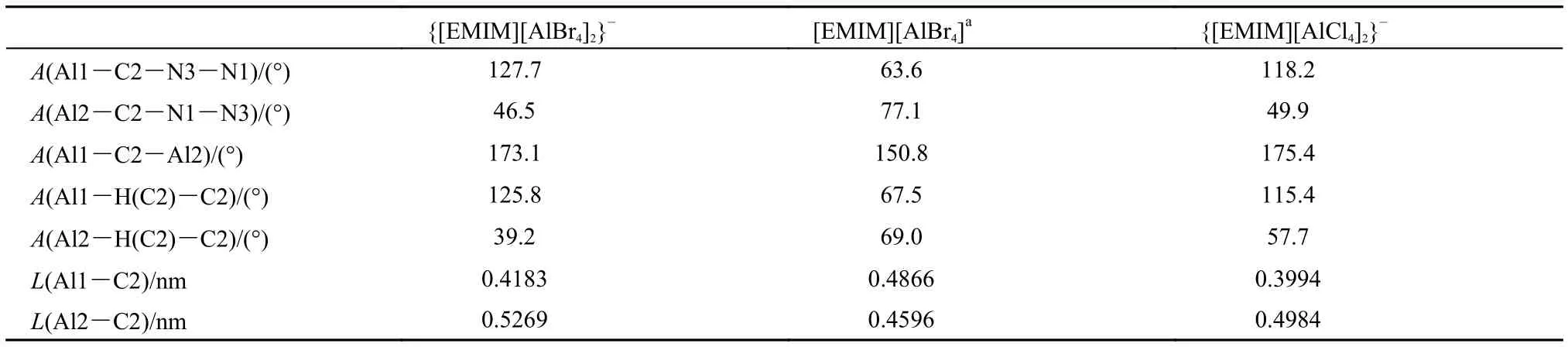
表4 溴鋁酸鹽晶體結構及{[EMIM][AlBr4]2}–, {[EMIM][AlCl4]2}–優化構型結構參數Table4 Structural parameters in [EMIM][AlBr4] crystal and optimized ion clusters of {[EMIM][AlBr4]2}–and {[EMIM][AlCl4]2}–
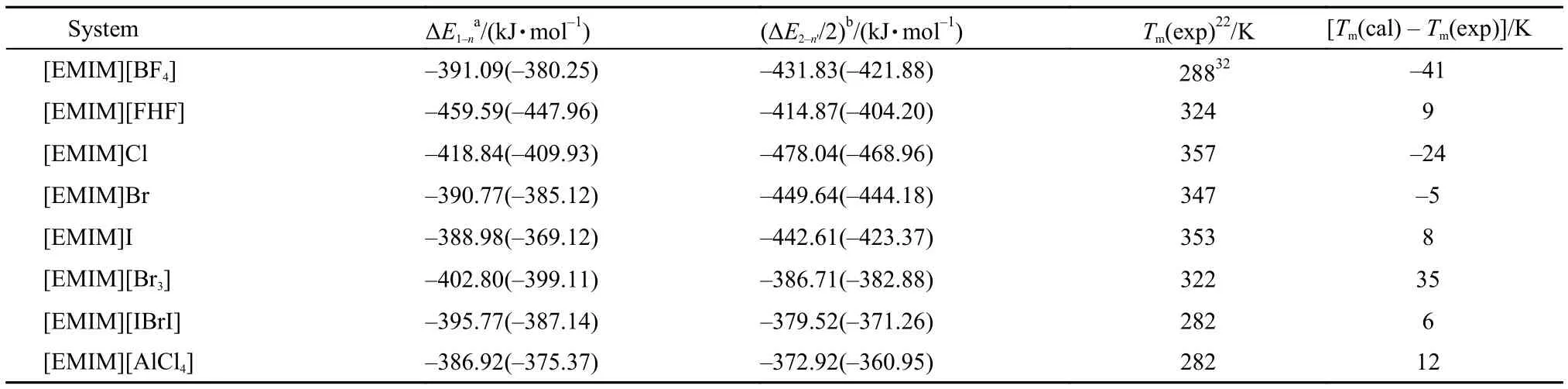
表5 離子簇模型中離子間相互作用能及實驗熔點Table5 Ion interaction energy and experimental melting points of ion cluster model
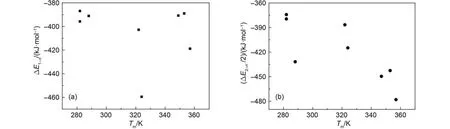
圖3 EMIM離子液體離子間相互作用能與實驗熔點之間的關系Fig.3 Relationships between the ion interaction energies and the experimental melting points of EMIM ion liquids
3.2 離子間相互作用能與實驗熔點之間的關系
所研究體系的{[EMIM]Xn}(n–1)–(X = Cl, Br, I, BF4, AlCl4, Br3, IBrI, FHF; n = 2, 3)和{[EMIM]2Xn'}(n'–2)–(n' = 3, 4, 5)模型的離子間相互作用能列于表5中(括號中數據為經過BSSE校正后的結果). 其中, ΔE1–n、ΔE2–n'分別代表{[EMIM]Xn}(n–1)–和{[EMIM]2Xn'}(n'–2)–模型的陰、陽離子間相互作用能. 從表5中數據可以看到除[EMIM]I體系外BSSE修正數值均較小. 不同體系的實驗熔點(主要來自文獻,22,32當有不同的熔點被報道時采用平均值)也列于表5中.
圖3更直觀地反映了實驗熔點與各離子簇模型離子間相互作用能ΔE1–n、ΔE2–n'/2 之間的關系. 可以看到對于陰離子不同的EMIM離子液體, 采用{[EMIM]2Xn'}(n'–2)–模型時實驗熔點與陰、陽離子間相互作用能之間趨向線性關系. 根據8個數據點線性擬合得到方程: Tm= –125.40 – 0.92091 × ΔE, 決定系數|R| = 0.74. 采用上述擬合方程預測得到的熔點值Tm(cal)與實驗值Tm(exp)的差值也列于表5中. 結果表明采用能夠反映晶體結構特征的多陽離子、多陰離子的離子簇模型, 陰、陽離子間相互作用能與實驗熔點存在定量關系.
4 結 論
對1-乙基-3-甲基咪唑鹵化物、氟硼酸鹽、三溴化物和二碘溴酸鹽、氯鋁酸和溴鋁酸鹽進行了離子簇模型研究, 所涉及的EMIM離子液體涵蓋了不同種類、不同尺寸陰離子的體系. 我們首先在B3LYP/6-311++G(d,p)水平上(對I使用6-311G(d,p)基組)分別對{[EMIM]2X4}2–(X = Cl, Br, I, BF4)、(X' = AlCl4, AlBr4, Br3, IBrI)、{[EMIM][FHF]3}2–、{[EMIM]2[FHF]5}3–共14種離子簇進行結構優化, 并對鹵化物和氟硼酸鹽進行了振動光譜計算. 結果表明對于所選定的體系, 多陽離子、多陰離子的離子簇模型能夠更好地反映相應晶體中的結構及陰、陽離子相互作用特征. 我們同時對陰、陽離子間相互作用能與實驗熔點之間的關系進行了研究, 發現采用2個咪唑陽離子和n個陰離子組成的模型時不同類型的EMIM離子液體晶體實驗熔點與陰、陽離子間相互作用能之間呈現近線性關系. 借助理論計算預言離子液體性質一直是人們的一個研究目標, 我們在之前離子液體熔點構效關系研究中發現, 若想獲得普適性強、形式簡單、物理意義明確的定量構效關系表達式需要以合理的結構模型為出發點. 本文的工作進一步證明了對于離子液體這種特殊的體系, 有可能利用離子簇模型模擬晶體中的結構特征, 并進一步通過離子間相互作用能預言實驗熔點. 我們希望通過我們的工作為離子液體的理性化設計提供有力的理論支持.
(1)Rogers, R. D.; Seddon, K. R. Science 2003, 302, 792. doi: 10.1126/science.1090313
(2)Li, R. X. Green Solvent—the Synthesis and Application of Ionic Liquids; Chemical In dustry Press: Beijing, 2004. [李汝雄. 綠色溶劑—離子液體的合成與應用. 北京: 化學工業出版社, 2004.]
(3)Zhang, S. J.; Lü, X. M. Ionic Liquids—from Fundamentals to Applications; Scientific Publish Ltd.: Beijing, 2006. [張鎖江, 呂興梅. 離子液體—從基礎研究到工業應用. 北京: 科學出版社, 2006.]
(4)Hallet, J. P.; Welton, T. Chem. Rev. 2011, 111, 3508. doi: 10.1021/cr1003248
(5)Li, W.; Wu, X. M.; Qi, C. S.; Rong, H.; Gong, L. F. J. Mol. Struct.: Theochem 2010, 942, 19. doi: 10.1016/j.theochem. 2009.11.027
(6)Li, W.; Qi, C. S.; Wu, X. M.; Rong, H.; Gong, L. F. Acta Phys. -Chim. Sin. 2011, 27, 2059. [李 巍, 戚傳松, 吳新民,榮 華, 龔良發. 物理化學學報, 2011, 27, 2059.] doi: 10.3866/PKU. WHXB20110914
(7)Li, W.; Qi, C. S.; Rong, H.; Wu, X. M.; Gong, L. F. Chem. Phys. Lett. 2012, 542, 26. doi: 10.1016/j.cplett.2012.05.072
(8)Wu, Y.; Zhang, T. T. J. Phys. Chem. A 2009, 113, 12995. doi: 10.1021/jp906465h
(9)Mohajeri, A.; Ashrafi, A. J. Phys. Chem. A 2011, 115, 6589. doi: 10.1021/jp1093965
(10)Turner, E. A.; Pye, C. C.; Singer, R. D. J. Phys. Chem. A 2003, 107, 2277. doi: 10.1021/jp021694w
(11)Katsyuba, S. A.; Zvereva, E. E.; Vidis, A.; Dyson, P. J. J. Phys. Chem. A 2007, 111, 352. doi: 10.1021/jp064610i
(12)Xiao, X.; Guo, M.; Pei, Y.; Zheng, Y. Spectrochimica Acta Part A 2011, 78, 1492.
(13)Rao, S. S.; Gejji, S. P. Computational and Theoretical Chemistry 2015, 1057, 24. doi: 10.1016/j.comptc.2015.01.012
(14)García, G.; Atilhan, M.; Aparicio, S. Chem. Phys. Lett. 2014, 610–611, 267.
(15)Katritzky, A. R.; Lomaka, A.; Petrukhin, R.; Jain, R.; Karelson, M.; Visser, A. E.; Rogers, R. D. J. Chem. Inf. Comput. Sci. 2002, 42, 71. doi: 10.1021/ci0100503
(16)Zhang, S. J.; Sun, N.; He, X. Z.; Lu, X. M.; Zhang, X. P. J. Phys. Chem. Ref. Data 2006, 35, 1475. doi: 10.1063/1.2204959
(17)Varnek, A.; Kireeva, N.; Tetko, I. V.; Baskin, I. I.; Solov’ev, V. P. J. Chem. Inf. Model. 2007, 47, 1111. doi: 10.1021/ci600493x
(18)Ren, Y. Y.; Qin, J.; Liu, H. X.; Yao, X. J.; Liu, M. C. QSAR Comb. Sci. 2009, 28, 1237. doi: 10.1002/qsar.v28:11/12
(19)Kowsari, M. H.; Alavi, S.; Najafi, B.; Gholizadeh, K.; Dehghanpisheh, E.; Ranjbar, F. Phys. Chem. Chem. Phys. 2011, 13, 8826. doi: 10.1039/c0cp02581j
(20)He, L.; Tao, G. H.; Parrish, D. A.; Shreeve, J. M. J. Phys. Chem. B 2009, 113, 15162. doi: 10.1021/jp905079e
(21)Rahman, M. B. A.; Jumbri, K.; Basri, M.; Abdulmalek, E.; Sirat, K.; Salleh, A. B. Molecules 2010, 15, 2388. doi: 10.3390/ molecules15042388
(22)The Reaxys Database, http://www.reaxys.com.
(23)Hu, S. Z.; Zhou, Z. H.; Cai, Q. R. Acta Phys. -Chim. Sin. 2003, 19, 1073. [胡盛志, 周朝暉, 蔡啟瑞. 物理化學學報, 2003, 19, 1073.] doi: 10.3866/PKU.WHXB20031118
(24)Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 03, Revision E.01; Gaussian Inc.: Wallingford, CT, 2004.
(25)Morrow, T. I.; Maginn, E. J. J. Phys. Chem. B 2002, 106, 12807. doi: 10.1021/jp0267003
(26)van Duijneveldt, F. B.; van Duijneveldt-van de Rijdt, J. G. C. M.; van Lenthe, J. H. Chem. Rev. 1994, 94, 1873. doi: 10.1021/ cr00031a007
(27)Jansen, H. B.; Ros, P. Chem. Phys. Lett. 1969, 3, 140. doi: 10.1016/0009-2614(69)80118-1
(28)Boys, S. F.; Bernardi, F. Mol. Phys. 1970, 19, 553. doi: 10.1080/00268977000101561
(29)Dymek, C. J.; Grossie, D. A.; Fratini, A. V. J. Mol. Struct. 1989, 213, 25. doi: 10.1016/0022-2860(89)85103-8
(30)Scott, A. P.; Radom, L. J. Phys. Chem. 1996, 100, 16502. doi: 10.1021/jp960976r
(31)Elaiwi, A.; Hitchcock, P. B.; Seddon, K. R.; Srinivasan, N.; Tan, Y. M.; Welton, T.; Zora, J. A. J. Chem. Soc. Dalton Trans. 1995, 3467.
(32)Wang, J.; Yang, X. Z.; Wu, S. D.; Li, G. S. Properties and Applications of Ionic Liquids; China Textile &Apparel Press: Beijing, 2006. [王 軍, 楊許召, 吳詩德, 李剛森. 離子液體的性能及應用. 北京: 中國紡織出版社, 2006.]
Quantum Chemistry Calculations of Ion Cluster Models of EMIM Ionic Liquids
LI Wei1,*ZHANG Jing2QI Chuan-Song1
(1College of Chemical Engineering, Beijing Institute of Petro-Chemical Technology, Beijing 102617, P. R. China;2College of Materials Science and Optoelectronics Technology, University of Chinese Academy of Sciences, Beijing 100049, P. R. China)
Different types of 1-ethyl-3-methylimidazolium (EMIM) ionic liquid compounds, including halides, tetrafluoroborate, tribromide, diiodobromate, chloroaluminate, and bromine aluminate, have been investigated using quantum chemical calculations. First, geometry optimizations of the ion systems, including {[EMIM]Xn}(n–1)–(X = Cl, Br, I, BF4, AlCl4, AlBr4, Br3, IBrI, FHF; n = 2, 3) and {[EMIM]2Xn'}(n’–2)–(n' = 3, 4, 5), were performed using the density functional theory (DFT) B3LYP method together with the 6-311++G(d,p) (6-311G(d,p) for I) basis set. The vibrational spectra were also calculated for the EMIM halides and tetrafluoroborate. The obtained structures and vibrational spectra were consistent with experimental results. In addition, a linear correlation between melting point and interaction energy was obtained for the {[EMIM]2Xn'}(n'–2)–models of the compounds studied.
Density functional theory; Imidazolium ionic liquid; Interaction energy; Ion cluster; Melting point
O641
10.3866/PKU.WHXB201507071
Received: March 30, 2015; Revised: July 6, 2015; Published on Web: July 7, 2015.
*Corresponding author. Email: liwei77@bipt.edu.cn; Tel: +86-10-81292127.
The project was supported by the Breeding Project of Outstanding Academic Leaders of Beijing Institute of Petro-Chemical Technology, China (BIPT-BPOAL-2014).
北京石油化工學院優秀學科帶頭人培育計劃(BIPT-BPOAL-2014)資助項目
? Editorial office of Acta Physico-Chimica Sinica
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
光學精密工程(2016年6期)2016-11-07 09:07:19
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
核科學與工程(2015年4期)2015-09-26 11:59:03
