外顯子組測序?qū)?例青壯年猝死的分子遺傳學(xué)分析
2015-12-13 12:26:32王純王輝許心舒許傳超賴小平陳銳林漢光邱升元
法醫(yī)學(xué)雜志 2015年6期
王純,王輝,許心舒,許傳超,賴小平,陳銳,林漢光,邱升元
(1.成都體育學(xué)院,四川成都610041;2.許昌市公安局刑事科學(xué)技術(shù)研究所,河南許昌 461002;3.廣州市刑事科學(xué)技術(shù)研究所,廣東廣州 510030;4.廣東醫(yī)學(xué)院,廣東東莞 523808)
外顯子組測序?qū)?例青壯年猝死的分子遺傳學(xué)分析
王純1,王輝2,許心舒3,許傳超4,賴小平4,陳銳4,林漢光4,邱升元4
(1.成都體育學(xué)院,四川成都610041;2.許昌市公安局刑事科學(xué)技術(shù)研究所,河南許昌 461002;3.廣州市刑事科學(xué)技術(shù)研究所,廣東廣州 510030;4.廣東醫(yī)學(xué)院,廣東東莞 523808)
目的以1例青壯年不明原因猝死綜合征(sudden unexplained death syndrome,SUDS)案例為研究對象,采用全外顯子組測序技術(shù),在全外顯子組水平尋找與SUDS相關(guān)的致病基因突變。方法對1例常規(guī)尸體解剖及病理學(xué)檢驗未發(fā)現(xiàn)明顯致死性病理改變的SUDS病例樣本,利用Ion Torrent PGMTM系統(tǒng)進行全外顯子組測序。測序數(shù)據(jù)以hg19為參照序列,并通過PhyloP、PolyPhen2、SIFT等軟件進行突變功能分析。最后設(shè)置三重條件過濾篩選有意義的單核苷酸變異:選取錯義突變-等位基因頻率<1%-蛋白質(zhì)功能預(yù)測。結(jié)果共發(fā)現(xiàn)4個罕見的可疑致病性單核苷酸變異。結(jié)合尸體解剖及病理學(xué)檢驗的結(jié)果,確定1個“高危害性”突變MYOM2(8_2054058_G/A)。PolyPhen2、SIFT的預(yù)測均為“有害”。結(jié)論利用二代測序技術(shù)進行全外顯子組水平的基因突變檢測和分析,可以為SUDS病例的死因分析提供新的方法和思路。MYOM2基因新可能是SUDS的致病基因,但其具體機制仍需進一步研究。
法醫(yī)遺傳學(xué);法醫(yī)病理學(xué);突變;不明原因猝死綜合征;全外顯子組測序
心源性猝死(sudden cardiac death,SCD)是目前最常見的死亡原因之一。據(jù)不完全統(tǒng)計,我國的SCD發(fā)病率約為41.8/100000[1],以此推算每年發(fā)生在我國的SCD病例約50萬。大多數(shù)SCD病例在隨后的解剖及病理檢驗中可發(fā)現(xiàn)冠心病、心肌炎等明確的病理改變。但有部分病例,在解剖及病理學(xué)檢查中,均不能發(fā)現(xiàn)明顯的致死性形態(tài)學(xué)改變,即解剖陰性的不明原因猝死(conventional autopsy-negative sudden unexplained death),這類病例多見于青壯年群體和兒童(0~35歲),因此也稱作青壯年不明原因猝死綜合征
(sudden unexplained death syndrome,SUDS)[2]。
近年來,研究者發(fā)現(xiàn)SUDS的發(fā)病特點與一些特殊類型的心臟疾病——離子通道病/心肌病(channelopathies/cardiomyopathies)[3-4]的特點相似,如患者通常表現(xiàn)“健康”,不知道自己的病情,心源性猝死常為首發(fā)癥狀。離子通道病/心肌病是多種疾病的總稱,包括長QT綜合征(long QT syndrome,LQTS)、Brugada綜合征(Brugada syndrome,Brugada S)、兒茶酚胺敏感性多形性室速(catecholaminergic polymorphic ventricular tachycardia,CPVT)等心肌離子通道病[5-7],以及肥厚型心肌病(hypertrophic cardiomyopathy,HCM)、擴張型心肌病(dilated cardiomyopathy,DCM)、致心律失常型右心室心肌病(arrthythmogenic right ventricular cardiomyopathy,ARVC)等心肌病[8-10]。隨著分子遺傳學(xué)研究的深入,研究者發(fā)現(xiàn)這類疾病都具有一定的遺傳背景,分子遺傳學(xué)檢測和分析成為對此類疾病進行明確診斷的必要手段。
與臨床診斷明確的各種離子通道病/心肌病不同,法醫(yī)面臨的SUDS常缺乏生前的臨床檢查資料,而常規(guī)的解剖和病理學(xué)檢驗亦難以發(fā)現(xiàn)明確的病理改變。因此,在無法從病理學(xué)層面明確死者的心肌病類型時,進行分子遺傳學(xué)檢測時也無法明確待檢測的目標(biāo)基因范圍。由于與SUDS相關(guān)的致病基因多達近百個[3,11],僅與HCM相關(guān)的致病基因就有數(shù)十個,涉及的突變類型多達上千種[12]。如果利用傳統(tǒng)分子生物學(xué)技術(shù)對這些基因進行突變篩查,無疑是個成本極高且費時費力的工作。因此,SUDS的遺傳學(xué)檢驗至今未能普及。
外顯子組測序技術(shù)[13]是以二代測序技術(shù)為平臺的高通量基因組分析技術(shù),利用外顯子序列特異性捕獲技術(shù)將基因組全部外顯子區(qū)域DNA捕捉并富集后進行的高通量測序分析,一輪檢測即可得到一份樣本外顯子組序列信息,有效數(shù)據(jù)可以涵蓋目標(biāo)區(qū)域95%以上的序列。隨著二代測序技術(shù)的成本逐漸降低[14-15],外顯子組測序有望成為最有效的SNP及基因突變的檢測手段。本研究利用外顯子組二代測序技術(shù)對1例SUDS進行遺傳學(xué)檢測,以期發(fā)現(xiàn)其可疑致病突變基因,為死因不明、解剖陰性案例的法醫(yī)病理學(xué)診斷探索新途徑。
1 材料與方法
1.1 研究對象
猝死病例1例,男性,19歲。運動后突然倒地死亡,尸體解剖未發(fā)現(xiàn)可以解釋其死亡原因的典型形態(tài)學(xué)改變,并通過問其親屬已排除家族遺傳病史。
1.2 解剖及病理學(xué)檢驗
死者經(jīng)系統(tǒng)的法醫(yī)學(xué)尸體解剖,并取心臟、大腦、肝、腎、肺等主要器官行組織病理學(xué)檢驗。取心血進行常見毒物和藥物檢驗。
1.3 外顯子組測序
1.3.1 基因組DNA提取
取尸體骨骼肌組織50mg,利用細胞/組織基因組DNA提取試劑盒(DP1901,北京百泰克公司),提取基因組DNA。
1.3.2 構(gòu)建樣本DNA的外顯子文庫
按照Ion Torrent PGMTM的TargetSeqTMExome Enrichment操作手冊程序,構(gòu)建樣本DNA的外顯子文庫,主要包括以下步驟:
(1)利用AMPure XP核酸純化試劑盒及Ion ShearTMPlus Enzyme MixⅡ?qū)μ崛〉幕蚪MDNA進行純化和片段化(180~220bp);
(2)利用Agencourt AMPure XP磁珠純化系統(tǒng)對片段化的DNA進行純化;
(3)對純化的片段化DNA進行質(zhì)檢(Agilent Bioanalyzer),質(zhì)檢合格后的DNA片段進行Adapters連接和缺口修復(fù)及純化;
(4)利用Ion TargetSeqTMExome試劑盒對純化的DNA片段庫進行PCR擴增,擴增產(chǎn)物經(jīng)磁珠純化;
(5)在Agilent 2100 Bioanalyzer平臺上,利用Agilent High Sensitivity DNA試劑盒對擴增的DNA庫產(chǎn)物進行質(zhì)控;
(6)外顯子靶序列片段的富集:利用Ion TargetSeq Exome Probe Pool、TargetSeq Hybridization Solution A及Hybridization Enhancer B等對通過質(zhì)控的DNA文庫進行外顯子靶序列片段的雜交捕獲,利用Ion TargetSeq Amplification試劑盒對捕獲富集到的外顯子文庫片段進行PCR擴增,產(chǎn)物進行磁珠純化;
(7)用Ion PITMTemplate OT2 200試劑盒(No. 4482286)準(zhǔn)備引物,檢測exome-enriched library的片段長度分布,進行高通量測序前的模板準(zhǔn)備(定量、稀釋);
(8)用Ion Torrent PGMTM系統(tǒng),以稀釋好的外顯子文庫為模板,進行高通量平行測序。
以上試劑盒均購自美國Life Technology公司。
1.4 生物信息學(xué)分析
將樣本外顯子測序數(shù)據(jù)與參照序列(hg19)比對,進行SNP檢測和注釋、InDel檢測與注釋等生物信息學(xué)分析。再將單核苷酸多態(tài)性(SNP)或單核苷酸變異(single nucleotide variants,SNV)等位點與千人基因組計劃、dbSNP等標(biāo)準(zhǔn)數(shù)據(jù)庫信息進行比對,確定各SNP或SNV的基因頻率。篩選出等位基因頻率<1%的SNV,進一步確定發(fā)生在猝死相關(guān)基因[3,12]范圍內(nèi)
的SNV。
運用在線、生物大分子功能預(yù)測軟件PolyPhen2和SIFT,以及基因序列保守性分析軟件PhyloP和Grantham對外顯子組測序結(jié)果中檢測到的基因突變進行編碼產(chǎn)物蛋白質(zhì)的功能預(yù)測分析。
利用sanger法對上述步驟篩選出的基因突變進行測序驗證。
2 結(jié)果
2.1 法醫(yī)病理學(xué)檢驗
全身主要器官未發(fā)現(xiàn)明顯致死性病理改變。心臟質(zhì)量272g,外膜光滑、表面血管淤血;左右冠狀動脈開口及主干無狹窄,主動脈根部內(nèi)膜光滑;各瓣膜及各瓣口周徑未見明顯異常;左心室壁厚1.4 cm,右心室壁厚0.4cm,室間隔厚1.2cm。鏡下:心外膜疏松,冠脈管壁未見明顯異常,大血管空虛,小血管淤血;心肌間隙彌漫性增寬、疏松,心肌纖維彌漫性波浪樣不規(guī)則扭曲、粗細不均,散在不規(guī)則心肌斷裂,偶見灶性出血,間質(zhì)血管淤血,未見明顯炎癥細胞浸潤。常見毒(藥)物檢驗均為陰性。
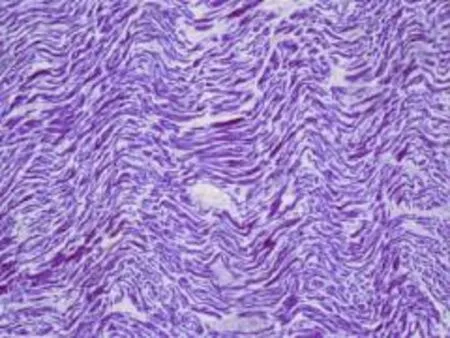
圖1 心肌彌漫波浪樣變,肌纖維斷裂HE×40
2.2 外顯子組測序
樣本的基因組DNA經(jīng)過外顯子組測序及隨后的生物信息學(xué)分析,在其基因組內(nèi)各個外顯子及其側(cè)翼部分區(qū)域內(nèi)共檢出24230個SNP位點,其中,新發(fā)現(xiàn)的SNV位點有590個。
鑒于SUDS病例的發(fā)病率極低,且很多為散發(fā)病例,因此,本研究將SNV作為可疑致病突變的篩查對象,測序結(jié)果見表1。經(jīng)過錯義突變-等位基因頻率<1%-蛋白質(zhì)功能預(yù)測三重篩選后,得到4個可疑致病突變位點信息(表2)。
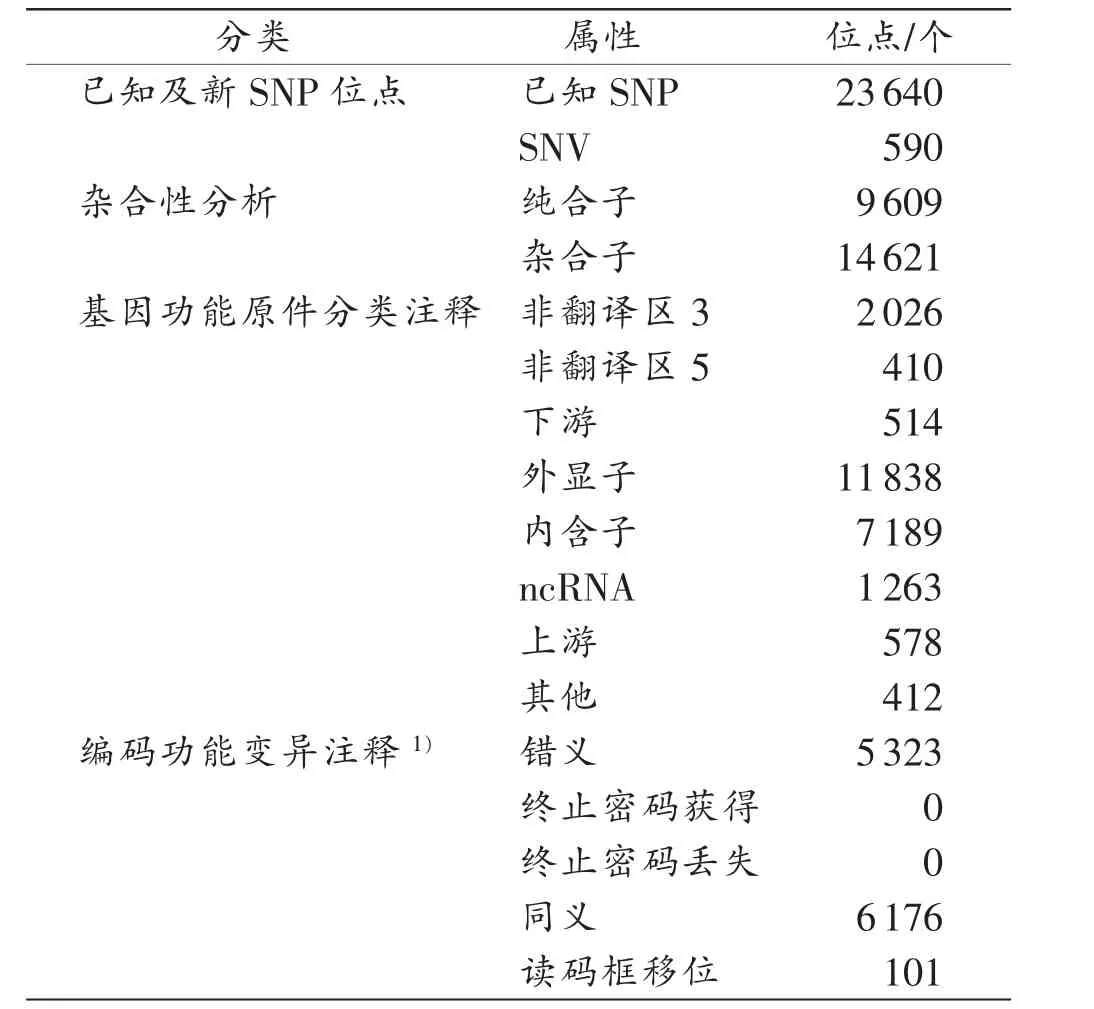
表1 樣本外顯子測序結(jié)果與數(shù)據(jù)庫外顯子及臨近側(cè)翼區(qū)域參考序列的比對

表2 檢測樣本在SUDS相關(guān)基因外顯子區(qū)域發(fā)現(xiàn)的部分SNV(MAF<1%)
2.3 sanger法測序驗證
對三重篩選后得到的4個SNV位點中兩個新發(fā)現(xiàn)的SNV位點進行sanger法測序驗證:
(1)8號染色體2054058位點(MYOM2基因)的G2761A的雜合突變;
(2)6號染色體7574921位點(DSP基因)的A2329G雜合突變。

圖2 Sanger法對外顯子組的驗證結(jié)果
3 討論
本例SUDS死者解剖及病理學(xué)檢驗未見各主要器官的致死性疾病和損傷,排除心肌炎、冠心病、先天性心臟病等基礎(chǔ)性疾病。心肌彌漫性不規(guī)則扭曲、斷裂及灶性出血符合急性心功能紊亂改變,腦、心、肺、肝、腎等全身器官高度淤血,符合急性心力衰竭致循環(huán)功能衰竭繼發(fā)改變。死者在疲勞狀態(tài)下突然倒地,迅速死亡,符合心源性猝死的發(fā)病特征。因此,在外顯子組測序后的遺傳學(xué)檢驗分析,主要的突變分析集中在與心血管系統(tǒng)結(jié)構(gòu)與功能相關(guān)的基因范圍內(nèi)[3,12]。
由于本研究檢測的樣本只有1例(家屬因心理壓力未能配合提供DNA樣本),在缺乏生前臨床資料以及了解到無家族遺傳背景的情況下,本研究設(shè)置了篩選SUDS相關(guān)的致病性SNV的條件:(1)選取處于91個基因[3,12]外顯子區(qū)域的錯義突變;(2)由于SUDS發(fā)病率低[2],可疑的致病突變應(yīng)為罕見突變,因此選取等位基因頻率<1%的SNV;(3)對第二步篩選到的突變進行dbSNP數(shù)據(jù)庫檢索及文獻檢索,選取新發(fā)現(xiàn)的突變和已經(jīng)報道為致病性的突變;對于新發(fā)現(xiàn)的突變,則需要通過軟件對其進行蛋白質(zhì)功能影響預(yù)測,如SIFT及PolyPhen2等。
本研究樣本中,外顯子區(qū)域共檢出兩萬多個SNV,在91個與心肌結(jié)構(gòu)和功能相關(guān)的基因范圍內(nèi),共篩選出錯義SNV 23個,主要分布于AKAP9、BAG3、CASQ2、DSP、JAG1、MYH7B、MYPN、MYOM1、MYOM2、RBM20、TTN、TXNRD2共12個基因的外顯子范圍內(nèi)。經(jīng)dbSNP數(shù)據(jù)庫信息篩選,排除已經(jīng)被證實為良性的突變,剩余12個SNV。依據(jù)MAF<1%的標(biāo)準(zhǔn)進行篩選,最終得到4個罕見SNV。其中,2個位點(分別位于AKAP9基因和MYH7B基因)為已在dbSNP數(shù)據(jù)庫注釋的SNP位點,2個為新發(fā)突變(分別位于DSP基因和MYOM2基因)。
第一個新發(fā)突變位于心肌細胞M線結(jié)構(gòu)的一種蛋白——M-protein(Myomesin 2)的編碼基因MYOM2中。MYOM2基因編碼的M-protein(Myomesin 2),相對分子質(zhì)量165000,由1465個氨基酸組成。是心肌細胞M帶的重要組成成分。通過與肌球蛋白、肌聯(lián)蛋白、輕酶解肌球蛋白等的結(jié)合發(fā)揮生理功能。本研究發(fā)現(xiàn)的MYOM2基因的22號外顯子的G2899A突變(rs370667622),使相應(yīng)的密碼子由GAT突變?yōu)锳AT,最終可以使合成的多肽鏈上發(fā)生Asp921Asn突變。這個突變位于M-protein多肽鏈高度保守的Ig-like C2-type 3區(qū)域。經(jīng)過SIFT、PolyPhen2的分析預(yù)測,該突變的結(jié)果為有害。根據(jù)以上預(yù)測結(jié)果,推測這個酸性氨基酸(Asp)被替換成中性(極性)氨基酸(Asn)的變異,將會影響M-protein與Myomesin、Titin等分子的親和力,這對心肌細胞肌小節(jié)結(jié)構(gòu)的穩(wěn)定性將產(chǎn)生顯著影響。在Rozanski等[16]的研究中發(fā)現(xiàn),心肌細胞的M-protein的表達受甲狀腺素T3的調(diào)控。T3對心肌細胞長期毒性效應(yīng)的分子機制是T3可下調(diào)心肌細胞M-protein的表達,進而嚴(yán)重影響心肌細胞的收縮速度。由此可見,M-protein的正常表達對于心肌細胞正常功能的維持具有關(guān)鍵作用。本研究發(fā)現(xiàn)的存在于SUDS病例的M-protein變異,有可能是該病例的死亡原因之一。該突變與心肌細胞功能障礙之間的相關(guān)性還有待進一步通過實驗來證實。
第二個新發(fā)突變是位于DSP基因的A2329G。DSP基因編碼橋粒斑蛋白,橋粒斑蛋白是心肌細胞間連接結(jié)構(gòu)-橋粒的主要成分,負責(zé)將中間絲錨定在橋粒斑,其N末端負責(zé)連接橋粒斑,C末端連接中間絲。DSP基因突變已經(jīng)被證實與心肌病ARVC相關(guān)[17-18]。本研究發(fā)現(xiàn)的DSP基因17號外顯子的錯義突變A2329G,將導(dǎo)致多肽鏈中的Ile777Val變異。經(jīng)過SIFT、PolyPhen2等軟件的預(yù)測分析,結(jié)果為良性(benign)。這提示該突變對DSP的轉(zhuǎn)錄產(chǎn)物橋粒斑蛋白的功能影響不大。本研究中的SUDS死者的右心室心肌也并未發(fā)現(xiàn)如右心室心肌層被纖維脂肪組織取代,心肌間質(zhì)脂肪浸潤等明顯的ARVC特征性病理改變,與軟件預(yù)測相符。
第三個低頻率SNV是AKAP9基因的C8935T(rs1063242,p.Pro2979Ser)。AKAP9基因產(chǎn)物Yotiao蛋白屬于A激酶錨定蛋白家族的一員。在交感神經(jīng)系統(tǒng)對心肌細胞動作電位時程進行調(diào)控的過程中,
AKAP9(Yotiao蛋白)可通過與KCNQ1鉀離子通道蛋白的α亞單位形成復(fù)合體而影響鉀離子電流Iks。KCNQ1基因突變可導(dǎo)致LQTS[19],Chen等[20]發(fā)現(xiàn)AKAP9分子的S1570L突變是一個LQTS家系的發(fā)病原因,該突變位于Yotiao蛋白與KCNQ1鉀離子通道蛋白結(jié)合的區(qū)域,進而影響Iks,導(dǎo)致LQTS的臨床體征。AKAP9基因的突變體與LQTS1相關(guān),本研究所發(fā)現(xiàn)的Pro2979Ser突變位于AKAP9分子C端,不在與鉀離子通道的結(jié)合區(qū)。PhyloP分析突變的位置并非序列保守區(qū)域,各種生物學(xué)分析結(jié)果均為“良性”。因此可初步判斷,AKAP9(Yotiao)蛋白的Pro2979Ser突變對KCNQ1鉀離子通道的Iks不產(chǎn)生明顯影響,因此本SUDS病例死于LQTS的可能性不大。
第四個低頻率SNV是RBM20。該基因編碼產(chǎn)物通過與RNA結(jié)合調(diào)節(jié)轉(zhuǎn)錄產(chǎn)物的剪接修飾,是具有多種功能的基因通過選擇性剪接表達出功能多樣的蛋白質(zhì)的重要調(diào)節(jié)蛋白。Refaat等[21]報道了RBM20參與心肌細胞肌節(jié)蛋白Titin的剪接調(diào)控,RBM20突變導(dǎo)致的剪接調(diào)控異常與DCM與相關(guān)。Brauch等[22]首先報道了在這個基因外顯子9區(qū)域內(nèi),在一段高度保守的富含精氨酸/絲氨酸的結(jié)構(gòu)域中發(fā)生的幾個突變(R636S、R636H和S637G)與家族性擴張型心肌病相關(guān)聯(lián)。Wells等[23]利用WES技術(shù)發(fā)現(xiàn)了RBM20基因的c.1907 G>A突變與DCM相關(guān)。在本例SUDS樣本中發(fā)現(xiàn)的RBM20基因c.2303G>C(p.Trp768Ser)突變雖然也位于9號外顯子,但突變位置(768位氨基酸)據(jù)Brauch報道的突變熱點區(qū)域相距100多個氨基酸,位于保守性較低的區(qū)域(PhyloP和Grantham評分均顯示此位點保守性較低)。該突變后果為色氨酸(Trp)突變?yōu)榻z氨酸(Ser),SIFT及PolyPhen2等預(yù)測軟件顯示該突變的危害性非常低。因此我們認為這個RBM20基因的c.2303G>C(p.Trp768Ser)不是本SUDS的主要致病突變。
此外,與Roncarati等[24]的研究提示相同,SUDS相關(guān)的各種心肌病有可能不是單基因病,其癥狀體征更可能是多個“有害”的多態(tài)性/突變基因協(xié)同作用的結(jié)果。這提示,全面檢測心肌細胞電生理功能及收縮舒張機械功能相關(guān)的各個基因,才能更加完整地解釋此類疾病的分子機制。
綜上所述,在本例SUDS樣本中,通過“等位基因頻率<1%的錯義突變-心肌細胞表達基因”的三步篩選,得到一個新發(fā)現(xiàn)的MYOM2基因的Asp921Asn突變,可能是其猝死的主要分子基礎(chǔ)。對于該突變對心肌細胞功能的影響,尚需進一步實驗來驗證。對于法醫(yī)學(xué)實踐中遇到的缺乏詳細生前臨床資料及家族遺傳病信息的猝死病例,外顯子組測序技術(shù)在鑒定致病突變及發(fā)現(xiàn)新突變領(lǐng)域具有不可替代的優(yōu)勢,是猝死案例遺傳學(xué)檢測的有力工具。
[1]Hua W,Zhang LF,Wu YF,et al.Incidence of sudden cardiac death in China:analysis of 4 regional populations[J].J Am Coll Cardiol,2009,54(12):1110-1118.
[2]Lim Z,Gibbs K,Potts JE,et al.A review of sudden unexpected death in the young in British Columbia[J]. Can J Cardiol,2010,26(1):22-26.
[3]Wilde AA,Behr ER.Genetic testing for inherited cardiac disease[J].Nat Rev Cardiol,2013,10(10):571-583.
[4]Ackerman MJ,Marcou CA,Tester DJ.Personalized medicine:genetic diagnosis for inherited cardiomyopathies/channelopathies[J].Rev Esp Cardiol(Engl Ed),2013,66(4):298-307.
[5]Goldenberg I,Moss AJ.Long QT syndrome[J].J Am Coll Cardiol,2008,51(24):2291-2300.
[6]Kaufman ES.Mechanisms and clinical management of inherited channelopathies:long QT syndrome,Brugada syndrome,catecholaminergic polymorphic ventricular tachycardia,and short QT syndrome[J].Heart Rhythm,2009,6(S8):S51-S55.
[7]Yl覿nen K,Poutanen T,Hiippala A,et al.Catecholaminergic polymorphic ventricular tachycardia[J]. Eur J Pediatr,2010,169(5):535-542.
[8]Mohiddin SA,Knight C.Interventional treatments for hypertrophic cardiomyopathy[J].Cardiovasc Ther,2012,30(3):e107-e114.
[9]McNally EM,Golbus JR,Puckelwartz MJ.Genetic mutations and mechanisms in dilated cardiomyopathy[J].J Clin Invest,2013,123(1):19-26.
[10]Marcus FI,Edson S,Towbin JA.Genetics of arrhythmogenic right ventricular cardiomyopathy:a practical guide for physicians[J].J Am Coll Cardiol,2013,61(19):1945-1948.
[11]Loporcaro CG,Tester DJ,Maleszewski JJ,et al. Confirmation of cause and manner of death via a comprehensive cardiac autopsy including whole exome next-generation sequencing[J].Arch Pathol Lab Med,2014,138(8):1083-1089.
[12]Efthimiadis GK,Pagourelias ED,Gossios T,et al. Hypertrophic cardiomyopathy in 2013:Current speculations and future perspectives[J].World J Cardiol,2014,6(2):26-37.
[13]Bick D,Dimmock D.Whole exome and whole genome sequencing[J].Curr Opin Pediatr,2011,23(6):594-600.
[14]Johansen Taber KA,Dickinson BD,Wilson M.The promise and challenges of next-generation genome sequencing for clinical care[J].JAMA Intern Med,2014,174(2):275-280.[15]Rabbani B,Tekin M,Mahdieh N.The promise of whole-exome sequencing in medical genetics[J].J Hum Genet,2014,59(1):5-15.
[16]Rozanski A,Takano AP,Kato PN,et al.M-protein is down-regulated in cardiac hypertrophy driven by thyroid hormone in rats[J].Mol Endocrinol,2013,27(12):2055-2065.
[17]Bauce B,Basso C,Rampazzo A,et al.Clinical profile of four families with arrhythmogenicright ventricular cardiomyopathy caused by dominant desmoplakin mutations[J].Eur Heart J,2005,26(16):1666-1675.
[18]den Haan AD,Tan BY,Zikusoka MN,et al.Comprehensivedesmosomemutationanalysisinnorth americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy[J].Circ Cardiovasc Genet,2009,2(5):428-435.
[19]Herbert E,Trusz-Gluza M,Moric E,et al.KCNQ1 gene mutations and the respective genotype-phenotype correlations in the long QT syndrome[J].Med Sci Monit,2002,8(10):RA240-RA248.
[20]Chen L,Marquardt ML,Tester DJ,et al.Mutation of an A-kinase-anchoring protein causes long-QT syndrome[J].Proc Natl Acad Sci U S A,2007,104(52):20990-20995.
[21]Refaat MM,Lubitz SA,Makino S,et al.Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy[J].Heart Rhythm,2012,9(3):390-396.
[22]Brauch KM,Karst ML,Herron KJ,et al.Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy[J].J Am Coll Cardiol,2009,54(10):930-941.
[23]Wells QS,Becker JR,Su YR,et al.Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy[J]. Circ Cardiovasc Genet,2013,6(4):317-326.
[24]Roncarati R,Viviani Anselmi C,Krawitz P,et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy[J].Eur J Hum Genet,2013,21(10):1105-1111.
(本文編輯:劉寧國)
Molecular Genetic Analysis of One Sudden Unexplained Death in the Young by Whole Exome Sequencing
WANG Chun1,WANG Hui2,XU Xin-shu3,XU Chuan-chao4,LAI Xiao-ping4,CHEN Rui4,LIN Hanguang4,QIU Sheng-yuan4
(1.Chengdu Sport University,Chengdu 610041,China;2.Institute of Criminal Science and Technology, Xuchang Public Security Bureau,Xuchang 461002,China;3.Guangzhou Institute of Criminal Science and Technology,Guangzhou 510030,China;4.Guangdong Medical College,Dongguan 523808,China)
Objective To find the mutation of disease-causing genes of sudden unexplained death syndrome(SUDS)in the young by whole exome sequencing in one case.Methods One SUDS case was found no obvious fatal pathological changes after conventional autopsy and pathological examination.The whole exome sequencing was performed with the Ion Torrent PGMTMSystem with hg19 as
equence for sequencing data.The functions of mutations were analyzed by PhyloP,PolyPhen2 and SIFT. A three-step bioinformatics filtering procedure was carried out to identify possible significative single nucleotide variation(SNV),which was missense mutation with allele frequency<1%of myocardial cell. Results Four rare suspicious pathogenic SNV were identified.Combined with the analysis of conventional autopsy and pathological examination,the mutation MYOM2(8_2054058_G/A)was assessed as highrisk deleterious mutation by PolyPhen2 and SIFT,respectively.Conclusion Based on the second generation sequencing technology,analysis of whole exome sequencing can be a new method for the death cause investigation of SUDS.The gene MYOM2 is a new candidate SUDS pathogenic gene for mechanism research.
forensic genetics;forensic pathology;mutation;sudden unexplained death syndrome;whole exome sequencing
DF795.2
A
10.3969/j.issn.1004-5619.2015.06.006
1004-5619(2015)06-0436-05
2011年四川省科技基礎(chǔ)條件平臺項目;廣東省科技計劃項目(2011B080701097)
王純(1961—),女,教授,主要從事運動性猝死和運動醫(yī)學(xué)研究;E-mail:993279039@qq.com
許傳超,男,博士,講師,主要從事分子遺傳學(xué)研究;E-mail:xcchao5855@sina.com
