心房結構重構在心房顫動的發生及維持中的作用
2015-12-16 05:59:23易茜綜述馬瑞彥審校
中國循環雜志 2015年8期
易茜綜述, 馬瑞彥審校
心房結構重構在心房顫動的發生及維持中的作用
易茜綜述, 馬瑞彥審校
心房顫動(房顫)在臨床上較為常見,具有致殘率和致死率高等特征,但其確切分子機制仍不明確。近年研究表明,心房重構是房顫發生、發展和維持的關鍵環節,而結構重構在其中的作用更為顯著。 本文綜述了腎素—血管緊張素—醛固酮系統、基質金屬蛋白酶系統、氧化應激、MicroRNA、內皮細胞間質轉化(EndMT)、遺傳基因多態性等參與結構重構的可能機制,為進一步探討房顫的發生和臨床治療提供了新的思路。
心房顫動; 心房重構; 結構重構;纖維化;分子機制
心房顫動(房顫)在臨床上較為常見,其發病率隨年齡增長而不斷上升[1],60歲以上人群發病率高達6% 以上[2],隨著全社會老齡人口的增多,房顫有向全球性疾病的趨勢發展[3]。其主要危害是腦卒中和肢體栓塞等并發癥,已成為心血管疾病發病和死亡的主要原因之一,對于房顫的治療、發病機制和臨床治療策略研究一直是心血管領域的重點。目前,房顫的治療主要包括內科抗血栓聚集、轉復竇性心律、減慢心室率等方面的藥物治療和射頻消融術,以及外科的改良迷宮手術(maze),但都存在很大的局限性,包括其治療的有效性以及術后出現的惡性心律失常、復發率高等[4],其根本原因是房顫的發生機制研究不透徹。故對房顫潛在分子機制的深入研究是提高臨床療效的前提。既往,對房顫的研究主要集中于電生理方面。近年來的研究表明,心肌膠原纖維的含量和構型改變是造成心臟重構的原因,結構重構在房顫中的作用和意義已被廣泛關注,本文就近年來關于結構重構的最新進展進行綜述。
1 房顫發生的基本機制
經典理論認為房顫的發生是心房內的異位點起搏以及微折返環的形成,多個異位灶相互碰撞形成折返,構成一個環路[5],這兩個基本機制目前已得到共識。房顫發生時,心房肌細胞快速激活,通過電生理重構,誘發快速性房性心律失常,心房內多地點的刺激,能夠產生空間的異質性,導致結構重構。研究表明,誘發房顫的始動因素和維持其病理狀態的基質是房顫相關機制的兩個主要部分[6]。目前認為房顫的發生和維持是多因素多通道共同作用的結果。
2 心房電重構
1995年Wijffels首次提出了“心房電重構”這一概念,通過建立快速心房起搏誘發山羊房顫模型發現:反復刺激導致心房有效不應期(AERP)縮短,與刺激時間成反比。刺激時間越長,房顫的誘發率越高,即是”房顫致房顫”理論。研究表明,快速房性心律失常可影響有效不應期的空間分布,增加空間異質性[7],成為觸發房顫的始動因素。其機制可能源于鈉、鉀通道的表達或功能異常,引起心肌電信號傳導紊亂,從而誘發心律失常。臨床證據也表明: 陣發性房顫患者隨著病程的延長,若不加以干預和控制,可發展為持續性房顫。近年來,人們認識到心房的結構重構和電生理重構是房顫發生和維持的重要基礎[8],盡管兩者在促房顫過程中的作用和意義不盡相同。
3 心房結構重構
Everett等[9]在犬的房顫模型上發現慢性房顫轉復為竇性心律后1~2周電生理改變完全恢復, 但心房的細胞形態和收縮功能卻不能同步恢復,這說明了影響房顫發生及維持的因素絕不是只有電重構。目前認為結構重構也可致局部心肌電活動傳導不均一,形成較多的微折返環,造成傳導阻滯;還可影響細胞間連接如縫隙連接蛋白(Cx)數量的缺失以及分布的改變[10],造成病變心臟的不協調收縮,進一步促進房顫的發展。
3.1 心房結構重構時心房肌細胞超微結構改變
心房結構重構主要表現在心肌細胞超微結構的改變、心肌間質膠原纖維重分布及纖維化等。光鏡和電鏡下可見:房顫動物模型和臨床標本90%以上心房肌細胞結構改變, 包括心房肌細胞肥大、溶解、肌纖維丟失、小而異形的線粒體數目增加、肌漿網的斷裂、細胞外基質增多、核周糖原堆積、α-肌球蛋白重鏈(MHC)向β-肌球蛋白重鏈轉化、α-平滑肌肌動蛋白(α-SMA)等胎兒期特征蛋白重新表達,稱為“反去分化( dedifferentiation)”; 心房肌加速合成的β-MHC 是胚胎型異構蛋白, 壽命短, 易衰竭, 加速了心肌重塑[11]心房肌結構的改變也給房顫的持續發生提供了一個良好的契機[12]。
3.2 參與心房結構重構的相關分子機制
房顫結構重構是心房纖顫的中心環節,心房纖維化是其主要表現,與其相關的分子機制較多,且各因素間的相互關聯錯綜復雜[13,14],圖1簡要概括了一些促房顫發生的因素,可望在分子水平揭示房顫本身的發病特點。
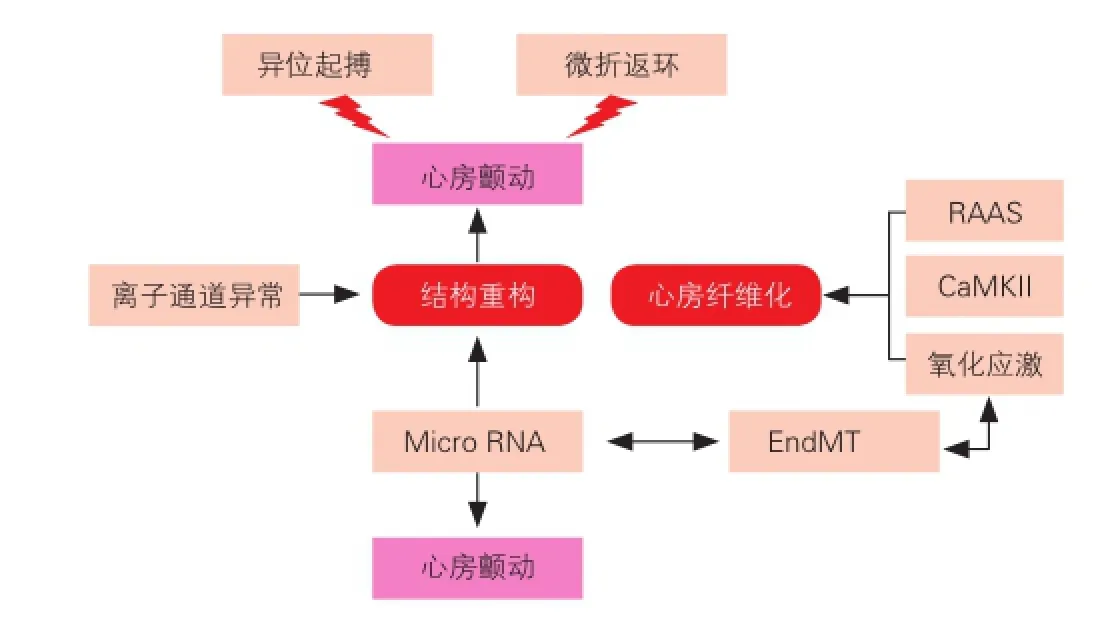
圖1 促心房顫動發生的因素
3.2.1 腎素—血管緊張素—醛固酮系統(RAAS)
RAAS系統在腎臟中的作用早已被廣泛研究,但對心房重構方面的影響在近幾年才開始被廣泛關注。房顫發生時,血管緊張素Ⅱ( AngII)生成增加,AngII可誘發心房纖維化,出現傳導阻滯、折返易于發生,而關于其致纖維化的機制,有學者認為其通過促分裂原激活蛋白激酶途徑誘發心肌間質細胞纖維網絡激活而導致心肌間質纖維化,也有研究認為[15],AngⅡ通過增強L-type 鈣通道的α1 C亞單位的微孔的表達來增強L-type鈣離子電流和轉運。另外AngⅡ可以增加心房的壓力,并通過增大心房內徑和改變心房的電生理重構而引發房顫。AngⅡ還能通過血管緊張素轉化酶1受體(AT-1)和AT-2 直接引起心房肌細胞凋亡,AngⅡ與心肌細胞共同孵育24 h能使凋亡細胞百分率較正常對照組增加5倍以上[16],可見AngII能通過多種途徑參與心房纖維化。近年來,醛固酮受體(MR)致心肌纖維化作用也日益受到重視[17],臨床上也早已有MR拮抗劑作為對抗纖維化的治療,代表性藥物如我們熟悉的螺內酯(安體舒通),在小鼠動物模型上已驗證增長的醛固酮水平能促進心房纖維化的進展,但有效性仍有待進一步研究。
3.2.2 基質金屬蛋白酶系統(MMPs)
MMPs 是一類降解膠原的最主要蛋白水解酶系統,它一方面介導膠原合成,促進間質纖維化,另一方面可促進胞間膠原分解斷裂,從而使心房結構疏松,發生間質重構,并主要以降解為主。MMPs與金屬蛋白酶組織抑制因子(TIMPs) 的平衡決定了細胞外基質( ECM)的含量[18]。新近研究證實房顫時心房肌MMPs表達增加,使正常的ECM 成分降解, 合成異常的膠原蛋白及結締組織, 參與心房結構重構。MMP-2、MMP-9是目前研究較多的明膠酶族[19]。Huxley等[20]發現房顫患者心房組織中MMP-9的活性升高, 且其活性與左心房直徑呈正相關, 表明MMP-9與房顫過程中的心房擴張及心房結構重構有相關性。Xu等[21]觀察心臟移植患者發現, 伴房顫者的心房肌較竇性心律者I型膠原容積分數明顯增加, 左心房增大,且MMP-2、MMP-9活性增高, 永久性房顫者更為突出;房顫患者TIMP-2蛋白水平下調;且心房MMP-2/TIMP-2比值與I型膠原容積分數呈正相關。上述研究結果初步提示, MMPs活性增高和TIMPs表達水平降低可能在房顫心房擴張和心肌重塑中起到了一定的作用, 二者之間的動態失衡會加速心肌重塑的進程。
3.2.3 炎癥和氧化應激(OS)
炎癥過程包括組織損傷、促炎因子釋放、炎癥細胞滲出、新生血管形成和瘢痕形成等環節。近年很多研究表明炎癥因子與許多器官(心、肺、腎、皮膚等)的纖維化聯系緊密[22,23]。氧化應激是指病理狀態下機體促氧化與抗氧化失衡時, 并產生過量活性氧自由基(ROS), 直接引起心肌細胞損傷和壞死, 導致心肌間質纖維化。ROS的促纖維化作用包括促成纖維細胞增加、向基質分生的肌原纖維轉變、促進前纖維基因表達的上調及引起MMPs/TIMPs失衡等。Lin等[24]采用熒光定量聚合酶鏈式反應(PCR)技術對14例慢性房顫患者右心耳線粒體研究發現,房顫患者線粒體DNA(mtDNA)氧化損傷程度較竇性心律者重, mtDNA氧化損傷最常見產物之一8-羥基-2脫氧鳥嘌呤核苷以及mtDNA含量較竇性心律者顯著增加, 故認為房顫患者心房肌 mtDNA存在氧化損傷。動物模型研究提示氧化應激相關蛋白可預測房顫發生。目前,ROS的促纖維化作用已被廣泛接受,但是其來源仍有爭議。研究者通過對煙酰胺腺嘌呤二核苷酸(NADPH)氧化酶缺乏的小鼠研究發現,其發生氧化應激的機率小于正常對照組,故認為NADPH氧化酶可能作為ROS來源參與了結構重構。
近來有關房顫的大量研究均提示多功能的鈣調素依賴蛋白激酶Ⅱ(CaMKII)能作為ROS的感受器,是發生心律失常前的一種信號[25],發生氧化應激時表達為ox-CaMKII,感受Ca2+濃度變化,增加了房顫的易感性。Purohit等[26]發現:注射了AngII的小鼠在經過快速心房刺激后房顫的易感性較對照組小鼠明顯增強。在臨床工作中使用藥物干預CaMKII轉化為ox-CaMKII可能作為一種新的治療靶點。
3.2.4 MicroRNA
MicroRNA(miRNA)是近來發現的一類高度保守、非編碼的、能在翻譯水平調節mRNA表達的小分子RNA。而最新研究發現miRNA 與心肌纖維化關系密切。miRNAs 能直接參與心肌纖維化,作用于心臟成纖維細胞,調控心臟成纖維細胞的存活[27],這些miRNAs在不同環節參與心房的結構重構,從而在房顫的發生與維持中發揮重要作用[28]。MicroRNA-29a 和-133a 在心肌中特異性表達與纖維化密切相關[29,30],Duisters等[31]研究發現miR-133過表達可使結締組織生長因子mRNA表達水平下降,減少心肌膠原蛋白的生成,抑制心肌纖維化,而且miR-133在人和鼠的胚胎、成人心臟及骨骼肌中均有表達,其表達水平和心肌肥厚存在負性相關。miR-29 表達于成纖維細胞中,調控mRNAs以編碼多種纖維蛋白、膠原蛋白、彈性蛋白等與心肌纖維化相關的蛋白。有研究者從mircroRNA 角度提出類似腫瘤發生學說中的“平衡”學說,即正常情況下,心臟中數百的microRNA 在調控靶基因時處于一種平衡狀態,而當某些因子介入后,打破了平衡,致使其調控的靶基因表達發生改變,導致心臟電重構及結構重構的發生,從而誘發房顫。
3.2.5 內皮細胞向間充質細胞的轉分化(EndMT)
EndMT是指病理或生理狀態下,內皮細胞在受到刺激因子的作用下,發生形態、結構和功能上的改變,具體表現為:內皮細胞的主要標志物血小板內皮細胞黏附分子(PECAM-1/ CD31)等表達減弱或消失,表達為成纖維細胞標志物如α-平滑肌肌動蛋白(α-SMA)、波形蛋白(vimentin)、成纖維細胞特異性蛋白-1(FSP-1,也被稱作S100A4)、I型和III型膠原蛋白等。有研究發現:小鼠心肌纖維化過程中有EndMT參與,并推測出約27%~35%的成纖維細胞來源于經EndMT轉化的內皮細胞,心肌纖維化程度減輕的同時常伴有EndMT減少[32]。體外培養人真皮層的微血管內皮細胞,給予炎癥因子刺激,能表現出成纖維細胞特性,表達出α-SMA、鈣調蛋白和Ⅰ型膠原蛋白等,提示真皮微血管內皮細胞在炎癥因子誘導下發生了EndMT[33]。Ghosh等[34]利用miRNA芯片技術檢測了轉運生長因子(TGF)-β誘導的內皮源性(發生了EndMT)成纖維細胞內miRNA水平,發現miRNA-125b、miRNA-21和miRNA-30b水平在EndMT過程中顯著升高,相反另外一些miRNA,如miRNA-122a、miRNA-127明顯降低。近年,對miRNA調節EndMT機制的研究已成為一大熱點,并取得了不少成果,可能為抑制病理狀態下EndMT,進而為抗心肌纖維化找到新的靶點。
3.2.6 遺傳基因多態性
人類基因組存在著約140多萬單核苷酸多態性(SNP),Ng報道人的基因組約有3000多個SNP,其中1/4是有害的,腎上腺素能受體(β2-AR)是心血管上重要的受體亞型,其基因多態性與高血壓、心力衰竭、心肌肥大、肥胖、血脂等病變密切相關。眾多研究也證明ACE兩種等位基因( I/D)多態性與左心室擴張及擴張性心肌病密切相關。 ACE I/D多態性決定了體內的ACE水平,釋放大量的RAAS,促進心房纖維化,參與結構重構[35]。心血管系統往往涉及涉及多個基因以及環境的相互作用,尤其大多數心血管疾病具有遺傳復雜性,根據基因型決定個性化治療方案是未來的一種理想趨勢,但目前因研究局限,仍有大量工作需要完成。
3.3 心房逆重構
近來,心房逆重構越來越得到重視,為預防和治療房顫提供了最新思路。逆重構(reverse remodeling)是指房顫轉復后心臟電生理特征及收縮功能得以恢復,組織結構去分化程度減輕。逆重構是個緩慢過程,尤以結構重構的逆轉更甚。Ausma等[36]研究房顫轉復后心肌溶解、肌漿網斷裂、心肌間連接組織等去分化改變逆轉,發現轉復后收縮功能恢復緩慢,可能是持續性房顫加劇了心房肌細胞的牽張,增加了Ca2+超載,心肌組織結構改變,延緩轉復后收縮重構的逆轉。另有研究證明,慢性AF患者在經過導管射頻消融術后,電重構恢復較快,心室率得到控制,但心臟結構重構作用并未停止,房室不同步及不規則的心室率可能在AF患者心臟結構重構中發揮重要作用[37]。Everett[9]等的動物模型研究發現,房顫轉復后4h即可觀察到逆重構改變,7~14天后心房有效不應期和房顫周期長度可完全恢復,但未觀察到結構重構恢復的改變。ACEI和ARB類藥物對于逆轉心臟重構有重要作用,其確切機制不明,可通過降低心房內壓力,抑制心房纖維化,改善心房內延遲傳導等,相關的RAS系統抑制劑藥物試驗仍在進行中。充分研究心房逆重構的過程和機制對于找到房顫治療的新靶點帶來了新的思路。
4 展望
綜上,房顫是一種復雜的心律失常,有多種調節機制及細胞內信號轉導途徑共同參與,各種機制間又存在交叉效應或共同通路,由于參與心房結構重構的因素和途徑的復雜性,可能對治療帶來一定難度,但阻止及逆轉心房結構重構可能成為未來治療房顫的新策略。雖然在多數患者中我們仍不能確定房顫的發生發展機制,在今后的基礎及臨床研究中,可能需要從更多角度開展研究以進一步了解房顫的發生發展機制,并進行相關的臨床干預以驗證治療效果。對心房結構重構相關分子機制的深入了解,可為房顫的臨床治療提供新的思路。
[1] Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation, 2006, 114: 119-125.
[2] Singh D, Cingolani E, Diamond GA, et al. Dronedarone for Atrial Fibrillation: Have we expanded the antiarrhythmic armamentarium . J Am Coll Cardiol, 2010, 55: 1569-1570.
[3] Wilke T, Groth A, Mueller S, et al. Incidence and prevalence of atrial fi-brillation: an analysis based on 8. 3 million patients . Europace, 2013, 15: 486-493.
[4] Nattel S. From guidelines to bench: implications of unresolved clinical issues for basic investigations of atrial fibrillation mechanisms. Can J Cardiol, 2011, 27: 19-26.
[5] 馬瑞彥, 肖穎彬. 心房顫動致心房重構的分子機制研究進展. 中國循環雜志, 2005, 4: 313-316.
[6] Miyagawa S, Sakaguchi T, Nishi H, et al. Recent clinical and experimental advances in atrial fibrillation. ISRN Cardiol, 2011, 2011: 1-12.
[7] Park JH, Pak HN, Lee S, et al. The clinical significance of the atrial subendocardial smooth muscle layer and cardiac myofibroblasts in human atrial tissue with valvular atrial fibrillation. Cardiovasc Pathol, 2013, 22: 58-64.
[8] Xu Y, Sharma D, Li G, et al. Atrial remodeling: New pathophysiological mechanism of atrial fibrillation. Med Hypotheses, 2013, 80: 53- 56.
[9] Everett TH , Li H , Mangrum JM , et al . Electrical morphological , and ultrastructural remodeling and reverse remodeling in a canine model of chronic atrial fibrillation . Circulation , 2000 , 102: 1454-1460.
[10] Igarashi T, Finet JE, Takeuchi A, et al. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation , 2012, 125: 216-225.
[11] 阮紅梅, 王瑋. 心房顫動發生機制的研究進展. 醫學綜述, 2011, 17: 37-40.
[12] Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol, 2008, 51: 802-809.
[13] Nattel S, Guasch E, Savelieva I. Early management of atrial fibrillation to prevent cardiovascular complications. Euro Heart J, 2014, 35: 1448-1456.
[14] Harada M1, Van Wagoner DR, Nattel S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ J, 2015, 79: 495-502.
[15] Tsai CT, Wang DL, Chen WP, et al. Angiotensin II increases expression of alpha1C subunit of L-type calcium channel through a reactive oxygen species and cAMP response element binding proteindependent pathway in HL-1 myocytes . Circ Res, 2007, 100 : 1476-1485.
[16] Von Lewinski D, Kockskamper J, Rubertus SU, et al. Direct proarrhythmogenic effects of angiotensin Ⅱ can be suppressed by AT1receptor blockade in human atrial myocardium . Eur J Heart Fail, 2008, 10: 1172-1176.
[17] Young MJ. Mechanisms of mineralocorticoid receptor-mediated cardiac fibrosis and vascular inflammation. Curr Opin Nephrol Hypertens, 2008, 17: 174-180.
[18] Kim SK, Park JH, Kim JY, et al.High plasma concentrations of transforming growth factor-β and tissue injibitor of metalloproteinase-1--potential non-Invasive predictors for electroanatomical remodeling of atrium in patients with non-valvular atrial fibrillation. Circ J, 2011, 75: 557-564.
[19] 董麗君, 湯寶鵬, 許國軍, 等. 心房肌基質金屬蛋白酶及其抑制劑、凋亡相關基因表達改變與增齡性心房顫動關系的研究. 中國循環雜志, 2012, 29: 1034-1038.
[20] Huxley RR, Lopez FL, MacLehose RF, et al. Novel Association between Plasma Matrix Metalloproteinase-9 and Risk of Incident Atrial Fibrillation in a Case-Cohort Study: The Atherosclerosis Risk in Communities Study. PLOS one, 2013, 8: 1-8.
[21] Xu J, Cui G, Esmailian F, et al. Atrial extracellular matrix remodeling and the main- tenance of atrial fibrillation . Circulation, 2004, 109: 363.
[22] Hashimoto N, Phan SH, Imaizumi K, et al. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol, 2010, 43: 161-172.
[23] Zeisberg EM, Potenta SE, Sugimoto H, et al. Fibroblasts in kidney fibrosis emerge via endothelial-to- mesenchymal transition. Am Soc Nephrol, 2008, 19: 2282-2287.
[24] Lin PH, Lee SH, Su CH, et al. Oxidative damage tomitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic BiolMed, 2003, 35 : 1310.
[25] Erickson JR, He BJ, Grumbach IM, et al. CaMKII in the Cardiovascular System: Sensing Redox States. NIH Public Access, 2011, 91: 889-915. [26] Purohit A, Rokita AG, Guan X, et al. Oxidized Ca2+/calmodulindependent protein kinase II triggers atrial fibrillation. Circulation, 2013, 128: 1748-1757.
[27] Lu Y, Zhang Y, Yang B, et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation, 2010, 122: 2378-2387.
[28] 何英杰, 郭玲, 丁振華. miRNA分子miR-24的研究進展. 解放軍醫學雜志, 2009, 34: 1145-1147.
[29] Chen S, Puthanveetil P, Feng B, et al.Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. Cell Mol Med, 2014, 18: 415-421.
[30] He Y, Huang C, Lin X, et al, Micro R NA-29 family, a crucial therapeutic target for fibrosis diseases . Biochimie, 2013, 95 : 1355-1359.
[31] Duisters RF, Tijsen AJ, Schroen B, et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res, 2009, 104: 170-178.
[32] Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-tomesenchymal transition contributes to cardiac fibrosis. Nat Med, 2007, 13: 952-961.
[33] 舒曉蓉, 聶如瓊, 謝雙倫. 內皮間質轉化和心肌纖維化. 中華心血管病雜志, 2014, 42: 797-800.
[34] Ghosh AK, Nagpal V, Covington JW, et al. Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): differential expression of microRNAs during EndMT. Cell Signal, 2012, 24: 1031-1036.
[35] 張玲, 桂慶軍, 彭建業. ACE基因多態性與心血管疾病相關性的研究進展. 中國心血管病研究, 2010, 8: 460-462.
[36] Ausma J, van der Velden HM, Allessie MA. Reverse structural and gap-junctional remodeling after prolonged atrial fibrillation in the goat. Circulation. 2003, 107: 2051-2058.
[37] 劉飛, 徐建, 嚴激. 射頻消融術對心肌病陣發性心房顫動患者心臟逆重構的影響. 安徽醫科大學學報, 2015, 50: 223-226.
2014-09-03)
(編輯:梅 平)
國家自然科學基金(81471408)
400037 重慶市,中國人民解放軍第三軍醫大學第二附屬醫院 心血管外科
易茜 碩士研究生 主要從事心臟疾病外科監護治療和心房顫動結構重構的分子機制方面的研究 Email: danlanruozi@163.com 通訊作者:馬瑞彥Email:maruiyan2008@hotmail.com
R541.4
A
1000-3614(2015)08-0813-04
10.3969/j.issn.1000-3614.2015.08.024
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
哲學評論(2021年2期)2021-08-22 01:53:34
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
中華詩詞(2019年7期)2019-11-25 01:43:04
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
文苑(2018年21期)2018-11-09 01:23:06
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
中國衛生(2015年9期)2015-11-10 03:11:12
現代企業(2015年9期)2015-02-28 18:56:50
