蔬菜中農藥殘留檢測方法的質量控制初探
2016-01-20 17:58:53孫建東黃劍飛徐培培曹海燕李華
南方農業·下旬 2015年12期
關鍵詞:質量控制
孫建東 黃劍飛 徐培培 曹海燕 李華

X摘 要 《NY/T761-2008蔬菜和水果中有機磷、有機氯、擬除蟲菊酯和氨基甲酸酯類農藥多種殘留的測定》這個檢測標準方法在農殘檢測中是應用最多的,該標準前處理簡單,回收率高,準確率高,但檢測人員在操作中會出現精密度差,回收率低或高的現象,如果在檢測過程中操作人員在關鍵步驟中得到有效的質量控制,必然會取得滿意的檢測結果。
關鍵詞 蔬菜;農藥殘留;檢測方法;質量控制
中圖分類號:S481.8 文獻標志碼:B 文章編號:1673-890X(2015)36--03
近年來,蔬菜質量安全越發成為人們關注的焦點。因而蔬菜的農藥殘留檢測工作變得十分重要,從事該工作的人員肩負的責任也變得更加重大,檢測過程中,蔬菜的品種和農藥殘留的品種繁多,有一個嚴格和科學的檢測質量控制程序,是得出準確和可靠農殘數據結果的前提和保證。本文已某次人員能力驗證為例,介紹江蘇省如東縣農產品質量檢測中心多年來在使用《NY/T761-2008》方法中總結的質量控制方法。
1 材料與方法
1.1 檢測樣品
本次實驗樣品為普通白菜樣品,由本中心質量負責人加入農藥標液,共4個蔬菜樣品,1個空白樣品,3個平行樣品。每個樣品均25 g。
1.2 儀器與試劑
氣相色譜(安捷倫7890A配備FPD檢測器,安捷倫6890N配備ECD檢測器,帶自動進樣器)、HHS421-4電熱恒溫水浴鍋(上海華聯環境試驗設備公司)、高速勻槳機(IKA T-25)、氮吹儀(Dri-BlockDB-3A)。主要試劑:乙腈(色譜純)、正已烷(色譜純)、丙酮(色譜純)和氯化鈉(分析純)。
1.3 實驗方法
采用《蔬菜和水果中有機磷、有機氯、擬除蟲菊酯和氨基甲酸酯類農藥多殘留檢測方法》(NY/T 761-2008)進行檢測。
1.4 標準溶液的配置
由農業部環境保護科研監測所提供,甲胺磷、甲拌磷、樂果、水胺硫磷、乙酰甲胺磷、氧樂果、毒死蜱、甲基對硫磷、對硫磷 、三唑磷、氯氰菊酯、氰戊菊酯、百菌清、三唑酮、甲氰菊酯、氯氟氰菊酯和異菌脲,質量濃度均為100 ?g/mL,有機磷的標液用丙酮稀釋至0.1~0.2 ?g/mL,有機氯的標液用正已烷稀釋至0.1~0.2 ?g/mL。
1.5 實驗條件
有機磷檢測條件色譜柱:DB-1701(30 m×0.32 mm×0.25 ?m);溫度:90℃保持0 min,以20 ℃速度上升到200 ℃,保持4 min,再以25 ℃上升到260 ℃,保持2.1 min。進樣體積:1 ?L;不分流進樣;檢測器200 ℃;進樣口溫度:200 ℃;氮氣流速:60 mL/min;柱壓:25 psi;空氣流速:100 mL/min;氫氣流速75 mL/min。
有機氯的檢測條件色譜柱:HP-5(30 m×0.32 mm×0.25 ?m);溫度:90 ℃保持0 min,以15 ℃速度上升到280 ℃,保持10.00 min。進樣體積:1 ?L;進樣口:250 ℃,不分流進樣;檢測器300 ℃;進樣口溫度:250℃,氮氣流速:60 mL/min;柱壓:25 psi。
2 檢測過程及結果
2.1 有機磷農殘前處理
準確稱取25.0 g樣品放人勻漿機中,加入50.0 mL乙腈,在勻漿機中高速勻漿2min后用濾紙過濾,濾液收集到裝有5~7 g氯化鈉的100 mL具塞量筒中,收集濾液40~50 mL,蓋上塞子,劇烈震蕩1min,在室溫下靜止10 min,使乙腈相和水相分層,吸取10.00 mL乙腈溶液,放入燒杯中,在80 ℃水浴鍋上加熱,并氮吹,蒸發近干。加入2.0 mL丙酮,蓋上鋁箔,用約3 mL丙酮分3次沖洗燒杯,并轉移至離心管。定容至5.0 mL,在旋渦混合器上混勻。過濾膜,移入2 mL樣品瓶中,上氣相測定。
2.2 有機氯農殘前處理
將弗羅里析柱依次用5.0 mL丙酮+正已烷(10+90)、5.0 mL正已烷預淋洗,條件化,倒入上述凈化溶液,用15mL刻度離心管接收洗脫液,用5 mL丙酮+正已烷(10+90)沖洗燒杯后淋洗弗羅里析柱,并重復一次,在水浴50 ℃條件下,氮吹至小于5 mL,用正已烷定容至5.0 mL,混勻,移入2 mL進樣瓶中,待測。
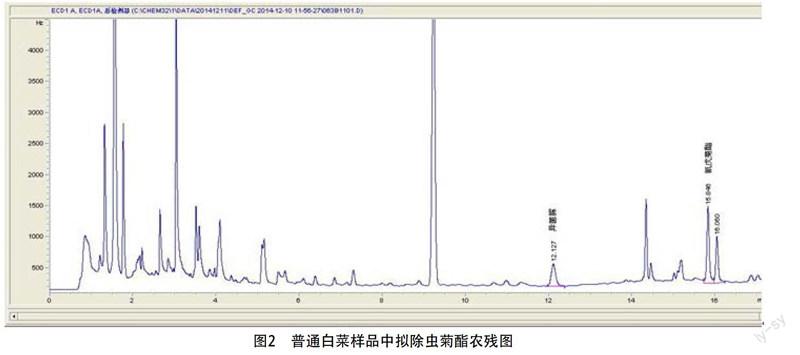
2.3 色譜圖(見圖1、圖2)
2.4 檢測結果
最后在樣品中檢測到甲拌磷、異菌脲和氰戊菊酯,定性準確,定量回收率在80%~110%,結果滿意。
3 檢測過程中的質量控制分析探討
3.1 檢測中耗材及標液配制過程中的質量控制
在農殘檢測準備工作中,要對所用的容量器具進行校驗,盡量使用色譜純試劑,并進行試劑空白試驗,確保無雜質峰出現。對于SPE小柱如弗羅里析柱等,要使用同一廠家的同一批次的產品,不同廠家不同批次的柱流速不同,并對于新購小柱進行回收率測定,經確認回收率正常方可使用;標準樣品一定要使用有證標準物質,并關注有效期。標準樣品儲存方法為-18~-20 ℃的冰箱中專人保管,有效期通常為1 a,配置上機使用的標樣時,不能直接稀釋計算,應先配置中間濃度的儲備液,如配成1 mg/L左右的多種農藥混合中間標樣,這樣既方便配置下一級標樣,又方便在0~5 ℃冰箱中保存,保存期為2~3周。
3.2 前處理過程中的質量控制
3.2.1 前處理提取過程中的質量控制
樣品稱樣進應先用樣品匙混勻樣品再稱樣,勻漿時勻漿機從低速中速到高速勻漿,并且每個速度時間一致,勻漿后過濾時,濾液倒入濾紙時要低于濾紙邊沿2 cm,防止濾液倒得太多,濺在外面,濾液要完全收集完。吸取10.00 mL乙腈提取液時,用10 mL大肚吸管,吸液前用該液潤洗3次,使液體充滿吸管內壁,吸取時刻度要準確。
3.2.2 濃縮凈化過程中的質量控制
氮吹時,氣體流速快慢要盡量一致,使平行樣氮吹濃縮條件一致;氮吹近干的要求是小燒杯內潮潮的一層,但不要吹干,吹干會回收率低,氮吹后加2 mL丙酮,一定要加蓋鋁箔,這一步在排風櫥中操作,如果不加蓋鋁箔,丙酮散發得快,會引起回收率高。在樣品多時,操作速度要快,定容完一個立即進行下一個,為防止時間過長,丙酮揮發。
凈化時淋洗弗羅里析柱時,所用淋洗液5 mL要準確,這樣可使弗羅里析柱的淋洗條件一致,液面不要干,當溶劑液面到達柱吸附層表面時,立即倒入上述待凈化溶液,如果柱子表面干了再倒入溶液,農藥會吸附在柱子上,回收率降低。第二次50 ℃氮吹時,要求氮吹至小于5 mL,可吹得小于1 mL,這樣做的好處是溶液中丙酮量會減少,用ECD檢測時不會對ECD有損害。
3.3 儀器使用過程中的質量控制
3.3.1 氣源檢查
如載氣(氮氣)、燃燒氣(氫氣、空氣)用鋼瓶,一般要選擇純度達99.99%,如用氣源發生器要經常檢查、更換干燥劑,注意氣源與儀器接口處是否有積水現象。最好配備氣體過濾器,避免鬼峰。
3.3.2 進樣器檢查
進樣器和進樣針要定期清潔并檢查進樣器是否靈活,以減少積塵太多造成停止運行或者進樣不重復性等。進樣針可以選用兩種不同的溶劑進行清洗;在進樣后增加清洗次數;時間長了可取下用丙酮浸泡清洗進樣針尾部等。
3.3.3 襯管檢查
襯管對殘留分析非常關鍵,有機磷農藥甲胺磷、氧化樂果、樂果等農藥的出峰情況受襯管影響非常大。最好選經惰性處理的襯管,當標樣峰面積有明顯下降時,要及時清洗或更換襯,更換后要獲得理想的響應值需要用樣品進行飽和。
3.3.4 色譜柱檢查
色譜柱是組分分離的重要部分,色譜柱質量如何直接影響檢測結果。衡量柱子好壞不僅看柱效、還要考慮其流失和柱壽命等。一般當標樣峰面積有明顯下降時,可以直接割去進樣口0.5~1 m的色譜柱,然后對柱子進行老化。
3.3.5 檢測器維護上需要區別對待
ECD的檢測池易污染,安捷倫儀器基線一般在500 Hz以下,1 000 Hz以上進需高溫過夜進行老化。FPD需調石英管的位置、氫氣流量等,安捷倫儀器基線一般在30pA以下,基線高一般要調節氫氣和空氣的配比。
3.3.6 樣品檢測前還要進行預實驗
氣相色譜儀要選擇最佳工作條件,使多種農藥能良好分離,峰型尖銳。一般還要重復進樣6針,計算峰面積,如RSD>10%,初步確定是襯管或柱子受污染,先從襯管排除,更換新襯管、O型圈和進樣墊,再次進樣品,確保RSD<5%,才可進行正常檢測。
3.4 加標回收的過程中的質量控制
大多數農藥的回收率會在70%~120%,但是氧化樂果等強極性農藥,有較強的基質效應,回收率會達160%以上,解決的辦法是同一樣品的基質配標液,例如找一空白樣品,按方法進行前處理,最后定容至5 mL,用這5 mL配制標液,進氣相測得峰面積,用來計算樣品回收率,能力驗證過程中也可用空白樣中多余的丙酮定容液配制標液,這樣就大大減少了基質效應。能力驗證中還要注意添加回收率試驗,在拿到盲樣進行前處理時,可自選一蔬菜樣品進行加標回收率試驗,蔬菜樣品盡量選擇與盲樣樣品基質一致的物質,加標后的樣品不能馬上進行前處理,等到樣品充分吸收農藥后方可進行,以保證加標回收試驗結果的準確性。
3.5 難檢組分的質量控制
難檢組分指檢測過程中添加回收太高或太低或回收率不穩定的一些農藥組分。按不同原因可分為以下幾類。
3.5.1 甲胺磷、氧化樂果、樂果
這些農藥的共同特點是極性相對較強,容易被襯管和玻璃棉吸附,一旦襯管臟了,標樣峰面積下降特別明顯。通過用樣品加標作為標液來計算,可以使它們的回收率變好。
3.5.2 甲拌磷
在做添加回收率時,甲拌磷的回收率不穩定,有時非常好90%左右,但有時會非常低,低于50%,而甲拌磷標樣出峰情況非常穩定,經過多次試驗發現是氮吹環節的問題,如果吹的太干,甲拌磷回收率肯定低,一定要近干,或者用旋轉蒸發儀進行濃縮就不會出現這個問題。
3.5.3 氰戊菊酯、氯氰菊酯
氰戊菊酯、氯氰菊酯也存在回收率不穩定情況,但往往回收率偏高,高到140%左右,這與基質效應有關,通過用樣品加標作為標液和自行加標回收來計算,可以使它們的回收率變高。
(責任編輯:劉昀)
猜你喜歡
中國科技博覽(2016年19期)2016-10-19 13:36:59
中國科技博覽(2016年18期)2016-10-19 11:06:33
中國科技博覽(2016年18期)2016-10-19 09:03:36
中國科技博覽(2016年18期)2016-10-19 08:46:18
科技視界(2016年21期)2016-10-17 17:58:28
中國實用醫藥(2016年24期)2016-10-17 06:28:30
科學與財富(2016年28期)2016-10-14 19:44:52
科學與財富(2016年28期)2016-10-14 18:58:41
科學與財富(2016年28期)2016-10-14 18:44:58
科技視界(2016年20期)2016-09-29 13:11:33
