
硫對鉑錸重整催化劑正庚烷脫氫環化性能的影響
2016-04-11 11:16:02張玉紅張大慶
石油煉制與化工 2016年12期
關鍵詞:催化劑
張 昭,張玉紅,張大慶
(中國石化石油化工科學研究院,北京 100083)
硫對鉑錸重整催化劑正庚烷脫氫環化性能的影響
張 昭,張玉紅,張大慶
(中國石化石油化工科學研究院,北京 100083)
分別采用常規還原硫化法、低溫注硫還原法、硫酸鹽浸漬法制備3種不同的含硫鉑錸重整催化劑,以正庚烷為模型化合物,進行3種催化劑的脫氫環化反應性能評價,并用TPR和吡啶吸附紅外光譜對催化劑進行表征,研究不同方式引入的硫對鉑錸重整催化劑正庚烷脫氫環化性能的影響。結果表明:與常規還原硫化法制備的催化劑相比,采用低溫注硫還原法制備的催化劑和硫酸鹽浸漬法制備的催化劑時具有更高的芳烴產率和選擇性,其中硫酸鹽浸漬法制備的催化劑上芳烴產率和選擇性最高。
重整 硫 正庚烷 脫氫環化
催化重整是石油煉制和石油化工的重要加工工藝之一,用于生產高辛烷值汽油調合組分和芳烴(BTX)[1-2]。按照操作方式不同分為連續重整和半再生重整,其中半再生重整使用的催化劑多為鉑錸體系雙功能催化劑[3]。硫對鉑錸重整催化劑的性能有著重要影響,它既是一種毒物,使催化劑發生中毒失活,但是也可以成為催化劑中的一個組元。鉑錸重整催化劑在使用前需經預硫化處理,以抑制催化劑中錸的強氫解性能[4],避免催化劑與原料接觸時發生強烈的放熱現象。在催化劑反應運轉過程中,硫是導致催化劑失活的毒物之一,主要來自于重整原料精制石腦油中,因此必須嚴格限制原料中的硫含量。在催化劑還原過程中,一般認為硫也是導致催化劑失活的毒物,對硫的控制應更為嚴格,要求在催化劑還原過程所用氫氣中H2S體積分數小于1 μLL[4]。然而,在前期研究中,與無硫H2氣氛下還原再硫化的常規方法相比,發現全過程含硫H2氣氛下還原催化劑具有更好的反應性能[5],同時還發現采用沉淀法合成的含有的氧化鋁為載體制備的催化劑也具有良好的反應性能[6]。然而,現有關于硫作用的認識尚不能對硫的這種促進作用給予合理的解釋。本研究通過不同制備方法引入不同硫物種,以正庚烷為模型化合物進行反應,考察不同催化劑的反應性能,研究硫物種對鉑錸重整催化劑上正庚烷脫氫環化反應的影響。同時,通過TPR表征方法研究催化劑中金屬的還原行為,通過吡啶吸附紅外光譜表征催化劑的酸性,初步探索硫物種對鉑錸重整催化劑影響的作用機理。
1 實 驗
1.1 催化劑制備
分別采用常規還原硫化法、低溫注硫還原法和硫酸鹽浸漬法3種不同方法引入硫物種制備催化劑。催化劑以中國石化催化劑長嶺分公司生產的條形γ-氧化鋁為載體,該載體由南非Sasol公司生產的高純SB粉擠條成型制得,標記為CL。
(1) 常規還原硫化法:以氯鉑酸、高錸酸銨為前軀體,與鹽酸配制成一定濃度的浸漬液,30 ℃下浸漬氧化鋁載體CL 3 h后70 ℃下真空干燥,然后在烘箱中120 ℃下干燥過夜,再置于管式爐中在空氣氣氛中500 ℃下活化4 h,得到氧化態催化劑。將氧化態催化劑置于管式爐中在H2氣氛中480 ℃下還原4 h后,然后降溫至425 ℃,切換為H2SH2混合氣對催化劑進行預硫化,所得常規還原硫化催化劑A。
(2) 低溫注硫還原法:將上述方法得到的氧化態催化劑置于管式爐中,在室溫下通入H2SH2混合氣,并在該氣氛下升溫至480 ℃,恒溫4 h,得到低溫注硫還原催化劑B。
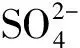
3種催化劑中Pt,Re,Cl,S元素的含量見表1。

表1 催化劑中Pt,Re,Cl,S元素含量 w,%
1.2 催化劑活性評價
催化劑的正庚烷脫氫環化反應在固定床微反裝置上進行。催化劑的裝填量為2 mL,反應在臨氫條件下進行,反應產物由在線氣相色譜進行定量分析。反應條件為:壓力(表壓)1.0 MPa,體積空速3 h-1,氫烴體積比800,反應溫度400~500 ℃。
原料的轉化率和產物產率采用面積歸一法計算(其中H2不計入產物中):


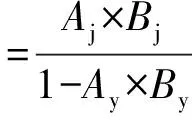
式中:Ai為i組分的色譜峰面積;Bi為i組分的相對質量校對因子;下標y和j分別表示原料和目標產物。
1.3 催化劑表征
TPR表征:在Micromeritics公司生產的ASAP2920動態吸附儀上進行。使用10%H2Ar混合氣,氣體流速為50 mLmin,升溫速率為10 ℃min。
酸性表征:采用Bruker公司生產的IFS113V-FT-IR光譜儀進行吡啶吸附紅外光譜分析。首先將樣品壓片,置于紅外光譜儀的原位池中密封,在350 ℃、10-3Pa下處理2 h后冷卻到室溫。導入2.67 Pa的吡啶蒸氣至原位池中,維持吸附平衡30 min后升溫到200 ℃,抽真空至10-3Pa,維持30 min,降至室溫攝譜。再升溫到350 ℃,抽真空至10-3Pa,維持30 min,降至室溫攝譜。
2 結果與討論
2.1 正庚烷脫氫環化反應性能
以正庚烷為原料,考察不同反應溫度下3種催化劑上正庚烷的脫氫環化反應性能,結果見圖1。從圖1可以看出:隨反應溫度的升高,3種催化劑上的正庚烷轉化率均明顯增加;在較低反應溫度時,各催化劑之間的正庚烷轉化率差別明顯,當反應溫度為400 ℃時,催化劑A,B,C上的正庚烷轉化率分別為12.69%,16.32%,29.07%,催化劑C上的正庚烷轉化率最高;隨著反應溫度的升高,不同催化劑上的正庚烷轉化率差距逐漸減少,在反應溫度為480 ℃和500 ℃時,不同催化劑上的正庚烷轉化率非常接近,反應溫度為500 ℃時,催化劑A,B,C上的正庚烷轉化率分別為96.41%,97.96%,95.79%,其中催化劑B上的正庚烷轉化率略高。

圖1 反應溫度對不同催化劑上正庚烷轉化率的影響■—催化劑A; ●—催化劑B; ▲—催化劑C。 圖2、圖3同
圖3為反應溫度對3種催化劑上的芳烴(包括甲苯和苯)產率的影響。從圖3可以看出:隨反應溫度的升高,產物中芳烴產率明顯上升;當反應溫度為500 ℃時,催化劑A,B,C上的芳烴產率分別為30.46%,33.93%,34.28%,低溫注硫處理獲得的催化劑B上芳烴產率比催化劑A上高3.47百分點,而采用CL載體制備的催化劑C上芳烴產率比催化劑A上高3.82百分點。

圖2 反應溫度對不同催化劑上C5+液體收率的影響
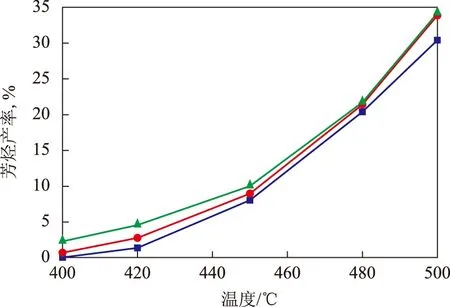
圖3 反應溫度對不同催化劑上芳烴產率的影響
表2為反應溫度為500 ℃時不同催化劑上正庚烷脫氫環化反應性能。從表2可以看出,相比催化劑A,采用催化劑B時C1~C2選擇性提高了0.19百分點,采用催化劑C時C1~C2的選擇性降低了0.39百分點,可以看出硫酸鹽浸漬法制備的催化劑C的氫解性能受到了一定程度的抑制。對于芳烴產率,采用催化劑B和催化劑C時都比采用常規還原硫化法制備的催化劑A時高,催化劑C上芳烴產率最高。比較3種催化劑上的芳烴選擇性,三者的差異更為明顯,采用催化劑B和C時分別比采用催化劑A時高3.05和4.20百分點,而采用催化劑C比B時高1.15百分點。綜上所述,當反應溫度為500 ℃時,對于正庚烷脫氫環化反應,相比常規還原硫化法制備的催化劑A,低溫注硫還原法制備的催化劑B和硫酸鹽浸漬法制備的催化劑C有更高的芳烴產率和選擇性,其中硫酸鹽浸漬法制備的催化劑C上芳烴選擇性最高。通過對比催化劑制備方法可知,催化劑B和催化劑C上的硫都是在催化劑上金屬未被還原時引入的,由此可知,在催化劑金屬未還原前引入硫有利于提高烷烴脫氫環化反應性能。

表2 500 ℃時不同催化劑上的正庚烷脫氫環化反應性能 w,%
Mills等[7]首先提出了在工業條件下各類重整反應的雙功能反應機理。雙功能反應機理考慮了2種催化中心的協同作用,對于脫氫環化反應來說,烷烴首先在金屬中心上脫氫生成烯烴(見圖4中反應1),然后烯烴在酸性中心上環化生成烷基環戊烷(反應2),再在金屬中心上脫氫生成烷烴環戊烯(反應3),再轉移到酸性中心上異構化為環己烯(反應4),然后由金屬中心催化脫氫為芳烴(反應5)。由于金屬中心上加氫-脫氫反應進行很快,酸性中心上的反應應該是雙功能反應中的控制步驟[4]。正庚烷脫氫環化反應機理如圖4所示。

圖4 正庚烷脫氫環化反應機理示意
從圖4可以看出,在雙功能反應機理中,金屬活性中心和酸性活性中心分別在反應過程中催化不同的反應,協同完成反應,金屬中心和酸性中心是影響催化劑反應性能的重要因素。因此考察硫物種對催化劑的金屬中心和酸性中心的影響是十分必要的。
2.2 TPR表征
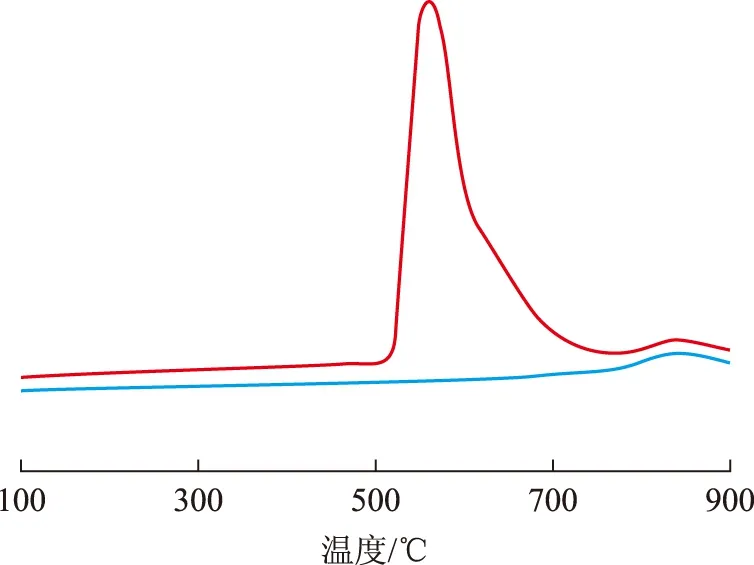
圖5 不同載體的TPR曲線 —CL載體; —CL載體
圖6為3種氧化態催化劑的TPR曲線。在催化劑A(O)的TPR曲線中,在200~400 ℃之間出現一個還原峰,在260 ℃左右達到峰值。這與很多學者[8-9]的研究結果一致,認為在鉑錸催化劑中鉑能夠催化錸的還原,鉑錸之間存在強的相互作用,該還原峰主要為鉑錸的共還原峰。
低溫注硫催化劑B(O)的還原基本上發生在150~500 ℃溫度范圍內,在200 ℃附近出現較強的還原峰,并在300 ℃附近出現一個肩峰,在500 ℃附近出現一個很小的還原峰;與催化劑A(O)的還原曲線相比,催化劑B(O)的主還原峰向低溫方向移動。崔少輝[10]的研究結果表明,150~500 ℃的還原峰由4個峰組成,峰位分別在200,220,300,500 ℃附近,其中200 ℃和500 ℃的還原峰歸屬為錸-硫的還原,300 ℃的還原峰歸屬為鉑-硫的還原,220 ℃的還原峰歸屬為鉑-錸-硫的共還原。由此表明在催化劑A(O)上鉑錸之間相互作用較強,鉑錸共還原程度較高;而催化劑B(O)上鉑錸之間的相互作用則相對較弱,鉑錸共還原程度較低,說明低溫注硫能減弱鉑錸之間的相互作用,減弱鉑錸共還原程度。
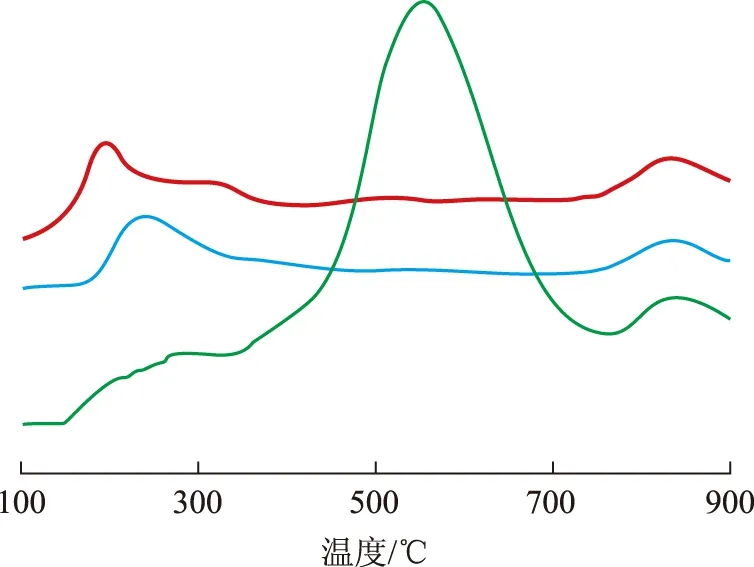
圖6 3種氧化態催化劑的TPR曲線 —催化劑A(O); —催化劑B(O); —催化劑C(O)
2.3 吡啶吸附紅外光譜表征
表3為3種催化劑的吡啶吸附紅外光譜表征結果。從表3可以看出:3種催化劑均只有L酸中心,沒有B酸中心;與催化劑A相比,催化劑B的強酸中心和弱酸中心酸量均顯著降低,催化劑C的強酸中心和弱酸中心酸量均明顯提高。


表3 3種催化劑的酸性結果
對比催化劑強酸量與弱酸量的比值發現,催化劑B和C的強酸量與弱酸量的比值均比催化劑A的高,尤其是催化劑C的這一比值明顯提高。Шипикин等[15]通過對比含有氯和氟的重整催化劑、因氯和氟大量流失而失活的催化劑、及經氯化處理的再生催化劑的反應性能,發現提高催化劑的酸性可以提高其烷烴脫氫環化反應的活性。因此催化劑B和C的這種酸性的調變有利于提高正庚烷在催化劑上脫氫環化反應活性及芳烴選擇性。
3 結 論
對3種催化劑的TPR和吡啶吸附紅外光譜表征結果表明,在催化劑上金屬未還原前引入硫可使催化劑的金屬變得更容易被還原,同時提高催化劑的強酸量與弱酸量的比值,有利于提高烷烴脫氫環化反應性能。與常規預硫化制備的催化劑相比,采用低溫注硫還原法和硫酸鹽浸漬法制備的催化劑具有更好的正庚烷脫氫環化反應性能,其中硫酸鹽浸漬法制備的催化劑不僅芳烴產率最高,而且芳烴選擇性也是3種催化劑中最佳的。
[1] 方大偉,馬愛增,張新寬.連續重整催化劑全生命周期技術經濟分析[J].石油煉制與化工,2015,46(12):1-4
[2] 孫建國.氯吸附技術在1.0 Mta連續重整裝置上的應用及其改造[J].石油煉制與化工,2014,45(6):74-78
[3] 張大慶,張玉紅,臧高山,等.半再生重整技術的現狀及發展[J].石油煉制與化工,2007,38(12):11-15
[4] 徐承恩.催化重整工藝與工程[M].北京:中國石化出版社,2004:182-537
[5] 崔少輝,張大慶,張玉紅,等.一種硫化態重整催化劑的制備方法:中國,CN103285895B[P].2015-11-25
[6] 張大慶,臧高山,張玉紅,等.一種石腦油重整催化劑及制備方法:中國,CN105363446A[P].2016-03-02
[7] Mills G A,Heinemann H,Milliken T H,et al.(Houdriforming reactions)Catalytic mechanism[J].Industrial & Engineering Chemistry,1953,45(1):134-137
[8] Wagstaff N.Alloy formation and metal oxide segregation in Pt-Reγ-Al2O3catalysts as investigated by temperature-programmedreduction[J].Journal of Catalysis:1979,59:434-445
[9] Isaacs B H,Eugene E P.The effect of drying temperature on the temperature-programmed reduction profile of a platinumrheniumalumina catalyst[J].Journal of Catalysis,1982,77:43-52
[10]崔少輝.鉑錸重整催化劑還原過程中硫的影響行為[D].北京:石油化工科學研究院,2012
[11]Arena F,Frusteri F,Mondello N,et al.Interaction pathway of chloride ions with γ-Al2O3:Surface acidity and thermal stability of the Clγ-Al2O3system[J].JCS Faraday Trans,1992,88(22):3353-3356
[12]Tanaka M,Ogasawara S.Infrared studies of the adsoption and the catalysis of hydrogen chloride on alumina and on silica[J].Journal of Catalysis:1970,16:157-163
[13]Imelik B.In catalysis by acids and bases[M].Elsevier:Amsterdam,1985:111-125

[15]Шипикин B B,Маслянский Г Н,и др.Прерващения углеводородов в присутствии отработанного и реактивировенного платиновых каталиэаторов реформинга[J].Нефтехимия,1966,6(3):401-406
EFFECT OF SULFUR ON DEHYDROCYCLIZATION OFn-HEPTANE OVER Pt-Re REFORMING CATALYSTS
Zhang Zhao, Zhang Yuhong, Zhang Daqing
(SINOPECResearchInstituteofPetroleumProcessing,Beijing100083)
Three different platinum-rhenium reforming catalysts were prepared by conventional reduction and presulfurization, sulfur injection at low temperature during reduction and the sulfate impregnation method, respectively. The performance of dehydrocyclization ofn-heptane over the three catalysts was investigated usingn-heptanes as a model compound. The catalysts were characterized by TPR and pyridine adsorption Fourier-transform infrared to test the effect of sulfur introduced by different ways on the performance of dehydrocyclization ofn-heptane. The results show that the catalysts prepared by sulfur injection at low temperature during reduction and the sulfate impregnation method exhibit better aromatics yields and selectivities compared to the catalyst prepared by conventional method. Especially the catalyst prepared by sulfate impregnation method reveals the highest aromatics yield and selectivity.
reforming; sulfur;n-heptane; dehydrocyclization
2016-05-12; 修改稿收到日期: 2016-07-12。
張昭,碩士,研究方向為催化重整和石油化工技術。
張玉紅,E-mail:zhangyh.ripp@sinopec.com。
中國石油化工股份有限公司合同項目(R15065-3)。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
