基于同步輻射的X射線成像技術在靜高壓研究中的應用*
2016-04-25 08:41:53侯琪玥敬秋民劉盛剛
高壓物理學報 2016年6期
關鍵詞:測量
侯琪玥,敬秋民,張 毅,劉盛剛,畢 延,柳 雷
(中國工程物理研究院流體物理研究所沖擊波物理與爆轟物理實驗室,四川綿陽 621999)
1 引 言
X射線成像技術伴隨著X射線的發現而誕生:在X射線發現之初,倫琴將手掌放在X射線管與熒光板之間,清晰地看到了手掌內部的骨骼結構,這是最早的X射線成像實驗,使人們意識到X射線在醫學領域有重要的應用前景。隨著同步輻射技術的發展,尤其是專用同步輻射裝置的出現,高能量、高準直性、高通量和高相干性的X射線光源成為現實。X射線成像技術也從最初的投影成像技術,發展出相襯成像、顯微成像、相干衍射成像等多種實驗技術[1-4]。物質的各種獨特性質與其結構息息相關,物質結構與性質之間的關系是凝聚態物理學、材料科學、結構生物學等學科的研究主線之一。高穿透性、無損傷的X射線成像技術可以在宏觀、介觀和微觀尺度提供物質結構的可視化信息,因此在眾多研究領域得到了廣泛的應用。
靜高壓實驗研究主要探討極端高壓環境下物質的結構和物性隨壓力和溫度加載的響應。壓力對物質的電子結構、聲子態密度、化學鍵、晶體結構,進而對材料的各種物性具有重要的調制作用。研究壓力對物質結構和物性的調制作用,可以促進人們對于相關科學問題的認識,如金屬絕緣體轉變、超導機制等;在高壓環境下,可以合成出具有優異力學或電學性能的材料,如超硬材料、熱電材料等[5-6];地球和行星內部是天然的極端高溫高壓環境,研究相關材料在高壓下的結構和物性對人類了解地球的形成、演化和發展具有重要的意義[7-8]。X射線衍射(X-Ray Diffraction,XRD)技術是最主要的材料結構表征技術之一,可以揭示具有長程周期性的晶體材料在原子尺度的結構信息;但是對于某些問題,XRD技術的應用具有一定的局限性,如不具備長程周期性的液體和無定形材料、介觀尺度問題等。而X射線成像技術對材料的周期性結構沒有要求,且能揭示材料從微觀、介觀到宏觀的跨尺度結構信息,因此在高壓研究中得到越來越廣泛的應用。
本文較系統地總結近年來X射線成像技術的發展及其在靜高壓研究中的應用,主要包括液體和無定形材料的密度測量,材料的相變動力學行為及鐵合金在地球內部的輸運機制研究等,并給出研究展望,以期對今后相關領域的研究工作有所幫助。
2 X射線成像原理
X射線投射到樣品后,一部分直接透射過去(即透射光),另一部分被一些微小結構(如包含物)以偏離入射方向散射出去(即散射光)。這些光會在樣品后的空間中進行衍射,根據傳播特點可分為幾何投影區、菲涅爾衍射區和夫瑯禾費衍射區,如圖1所示。在距離樣品很近的空間內,光幾乎為直線傳播,因此光屏上顯示為幾何投影,在該區域內的X射線成像為投影成像,空間分辨率主要取決于探測器的像素尺寸(從1微米到幾十微米),這種成像方法是傳統的吸收成像,旋轉樣品還可以進行三維重建。當光傳播到菲涅爾衍射區時,菲涅爾衍射效應開始增強,樣品的相位信息轉換為光強信息。某些相襯成像技術需要利用特殊的光學元件將相位信息提取出來,目前世界上較為常見的是光柵剪切相襯技術和衍射增強相襯技術。而同軸衍射成像技術只需要采集不同距離下的衍射圖像便可提取出相位信息。在此區域若利用菲涅爾波帶片對樣品進行放大成像,即為X射線透射顯微成像(Transmission X-Ray Microscopy,TXM)技術,其空間分辨率主要受物鏡的數值孔徑限制,一般為30~100 nm。當光傳播至夫瑯禾費衍射區域時,光屏呈現的是樣品的傅里葉變換散射圖,可根據散射圖反推樣品的電子密度,即X射線相干衍射成像(Coherent Diffraction Imaging,CDI)技術。 對于晶體樣品,需要采集晶體在某個衍射方向(hkl)下的衍射斑,即Bragg-CDI成像。這種成像技術的分辨率受最大散射角的限制,目前可達到10 nm[9],是X射線成像方法中分辨率最高的一種(見表1)。
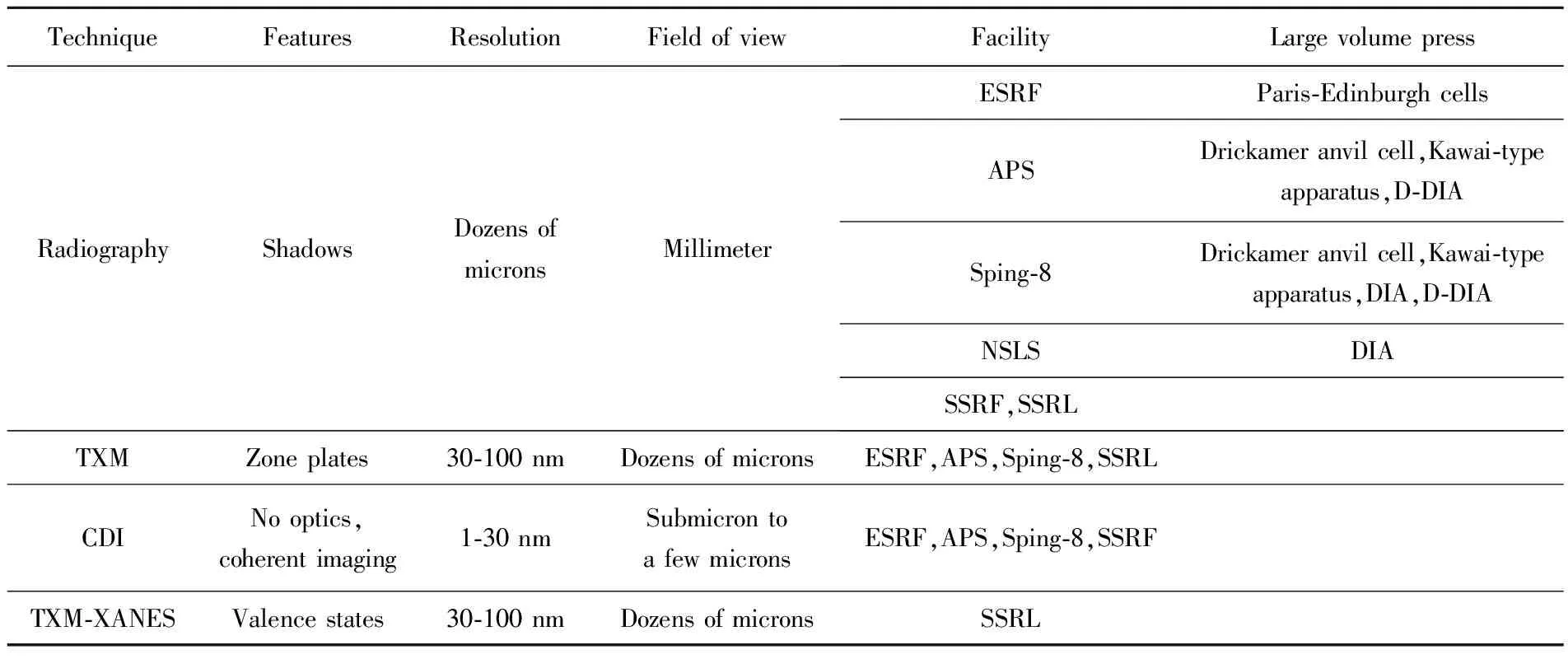
表1 典型的X射線成像技術及其技術特點Table 1 Typical X-ray imaging techniques and their properties
Notes:(1) Information concerning the synchrotron radiation facilities listed here is accessible online;the large volume presses are currently used in the corresponding beam-line;(2) NSLS:National Synchrotron Light Source at the Brookhaven National Laboratory;D-DIA:A hybrid system using a set of large DIA anvils to compress the Kawai cell;ESRF:European Synchrotron Radiation Facility;APS:Advanced Photon Source;SSRF:Shanghai Synchrotron Radiation Facility;SSRL:Stanford Synchrotron Radiation Lightsource; (3) XANES:X-Ray Absorption Near-Edge Spectrum.
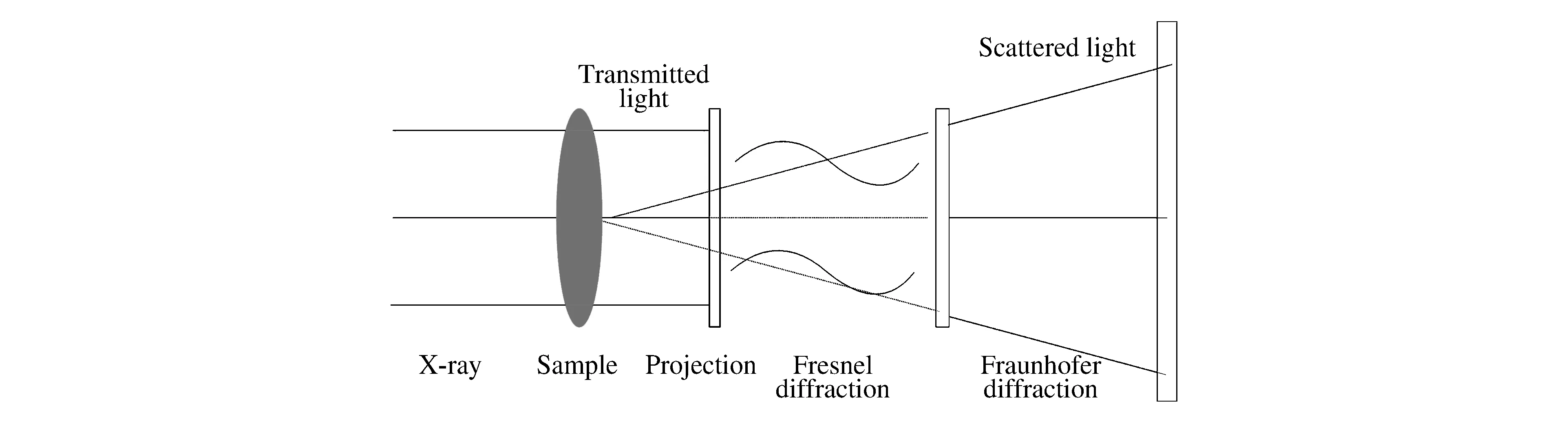
圖1 X射線成像的衍射區劃分Fig.1 Diffraction zones of the X-ray imaging
3 X射線成像技術在高壓研究中的應用
在高壓研究中,X射線成像技術在許多方面都發揮著無可替代的作用,如無定形材料的物態方程測量、高壓加載下的聲速測量、地球物理中熔融鐵在巖石中的輸運機理研究、晶體材料中的應變分布以及材料相變過程的演化研究等。以下將簡要介紹X射線成像技術在相關領域的研究進展。
3.1 物態方程測量
物態方程(Equation of State,EOS)是高壓實驗研究中最重要的研究內容之一,研究物質體積(密度)對壓力和/或溫度的響應,是物質性質的最本質描述之一。通常情況下,通過高壓(大壓機、金剛石對頂砧加載等)、高溫(電阻加溫、激光雙面加溫等)加載下的原位XRD實驗獲得材料的EOS。其中大壓機設備龐大,且不易搬運,表1列出了不同類型的大壓機在國內外同步輻射光源束線下的使用情況。XRD實驗適用于測量晶體材料的EOS,而對于非晶材料(如玻璃、液體和熔融態物質等),由于難以建立衍射信號與實空間樣品體積的簡單對應關系,使得利用衍射技術測量非晶材料的EOS具有很大挑戰。X射線成像技術則不受樣品狀態的限制,是測量非晶材料EOS的有效工具。
目前,用X射線成像技術測量EOS有兩種方式:密度測量和體積測量。最初的測量是從大壓機加載下樣品的密度測量開始[10-13]:沿徑向掃描樣品,得到圓柱形樣品對X射線的吸收強度隨徑向位置的變化,通過擬合得到樣品的密度。對于液態樣品,主要通過大壓機中的圓柱形樣品腔(一般為單晶藍寶石或氮化硼)維持樣品的幾何形狀。一維掃描方法對柱狀樣品形狀的要求嚴格,如果樣品發生形變,那么擬合出的密度參數會有很大的偏差。Chen等人[14]在2014年提出了X射線投影成像方法以擬合密度參數,該方法只要求在X射線的傳播方向上保持樣品形狀,在垂直于光傳播方向上的樣品變形對測量結果的影響很小(見圖2(a))。投影成像方法并非擬合柱狀樣品的吸收曲線,而是擬合樣品內紅寶石小球的吸收曲面(圖2(b)所示為樣品和紅寶石球的投影像),這種二維擬合方法可以減小一維擬合的統計誤差。上述擬合方法主要針對大壓機的測量。采用金剛石壓機加載時,要想通過樣品的吸收強度求出其密度參數[15],需要測量樣品厚度,而樣品厚度一般通過擬合封墊的一維厚度分布得到;封墊厚度的測量則需要結合XRD和縱向吸收掃描兩種技術,并且在計算中不考慮金剛石變形對封墊厚度的影響,此時可采用徑向斷層掃描成像技術直接測量樣品密度。徑向斷層掃描成像技術多用來測量體積參數,但是如果分辨率不夠高,或噪音的差別較大,則很難確定樣品邊界。2008年,Liu等人[16]直接在重建樣品內選擇明確的區域(如圖2(c)方框所示)進行密度測量。隨后,該密度測量方法的理論部分被Xiao等人[17]進行拓展:由于金剛石對頂砧(Diamond Anvil Cell,DAC)下的角度缺失會影響傳統的濾波反投影三維重建,參考物重建可以消除上述問題造成的系統誤差,如果參考物與樣品的EOS曲線趨勢相近,還可以進一步消除樣品的誤差震蕩。

圖2 X射線投影技術測量密度示意圖[14](a),X射線投影成像圖[14](b),以及金剛石壓機加載下的樣品(Pt、NaCl和Fe)、紅寶石球和傳壓介質[17](c)Fig.2 (a) Density measurement using X-ray radiography imaging[14];(b) simulated X-ray radiography imaging[14]; (c) the sample (Pt,NaCl,Fe),ruby ball and the pressure medium in the diamond anvil cell[17]
對于體積測量,最初也是在大壓機中實現的。2005年Wang等人[18]在美國先進光源(Advanced Photon Source,APS)的GSECARS實驗站上對Drickamer壓砧加載下(壓力小于8 GPa)硫化鐵基質中的Mg2SiO4小球(直徑0.8 mm)進行三維重建成像(分辨率為10 μm),證明了X射線斷層掃描的體積測量在大壓機中的適用性(見圖3)。隨著成像技術的發展,2012年Wang等人[19]利用全景DAC和APS 32 ID-C線站上的TXM技術,對4.7、8.1和12.0 GPa壓力下的晶體Sn顆粒(粒徑約10 μm)進行三維成像(分辨率達到60 nm),經三維重建和邊界分割后,計算出樣品的體積V(相對誤差為0.5%),所得的p-V曲線與之前XRD的測量結果非常吻合(見圖4)。由此可見,無論是大壓機加載還是DAC加載,X射線成像技術均能給出精確的體積測量結果。
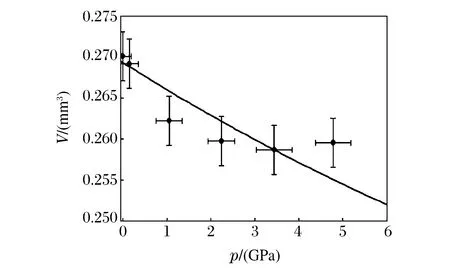
圖3 Mg2SiO4的p-V曲線[18]Fig.3 p-V curve of Mg2SiO4[18]
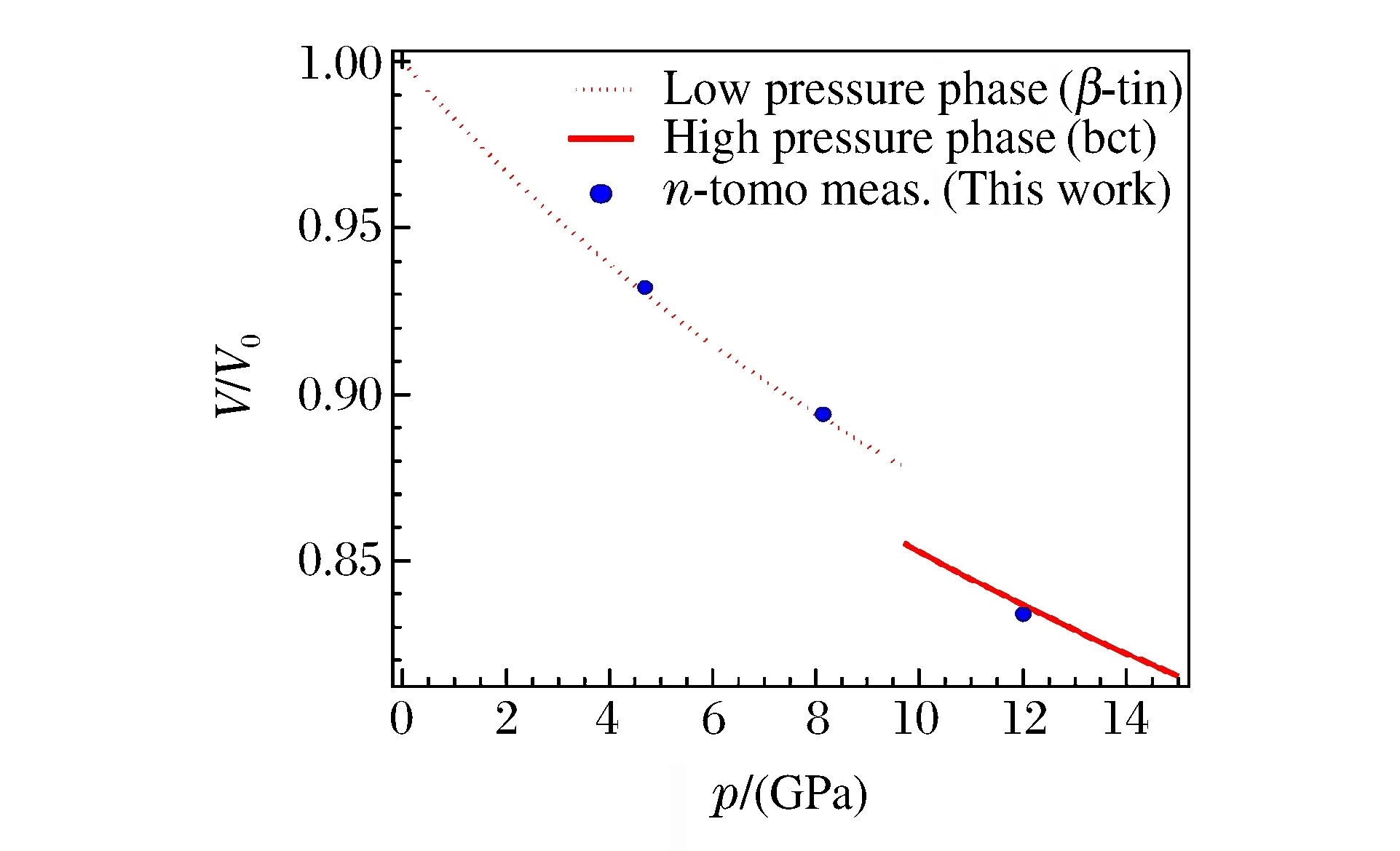
圖4 晶體Sn的p-V曲線[19]Fig.4 p-V curve of crystalline Sn[19]
上述測量密度和體積的X射線成像方法對于無定形材料及液體物態方程研究的意義重大。無定形材料的原子排列結構非常復雜,其XRD峰寬是晶體的10倍。盡管如此,無定形材料的第一衍射峰位Q1仍可用來描述“單胞”尺度d1(最近鄰原子間的平均尺寸),即d1∝1/Q1。通常認為,在無序各向同性系統中可用V∝(1/Q1)3估算體積[20-21]。但是,最近發現在“無序”的金屬玻璃中在短程范圍內存在著非常復雜的有序性[22-23],而Meade等人[24]利用此關系計算玻璃的p-V關系時,發現與理論預測值相差3倍,說明無定形材料的復雜原子結構特點使XRD技術受到限制。
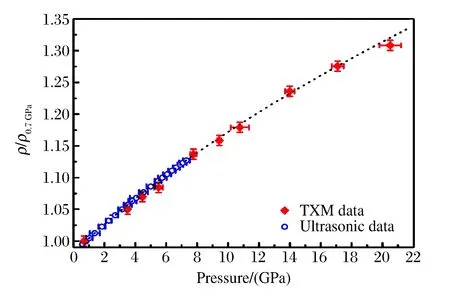
圖5 金屬玻璃的p-ρ/ρ0.7 GPa曲線[25]Fig.5 p-ρ/ρ0.7 GPa curve of metallic glass[25]
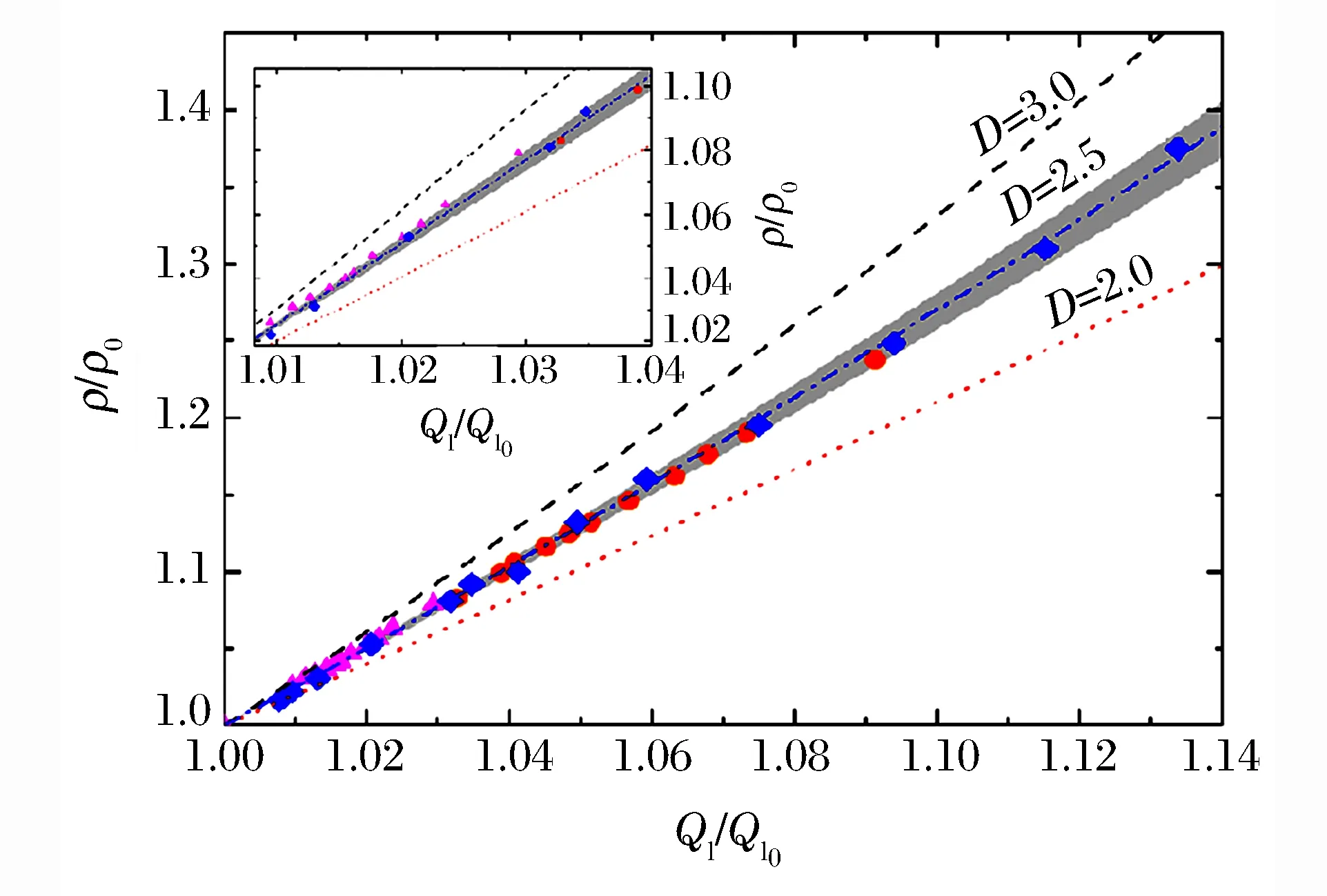
圖6 金屬玻璃的密度與第一衍射峰位的關系[25]Fig.6 Density vs.the position of the first diffraction peak of the metallic glass[25]
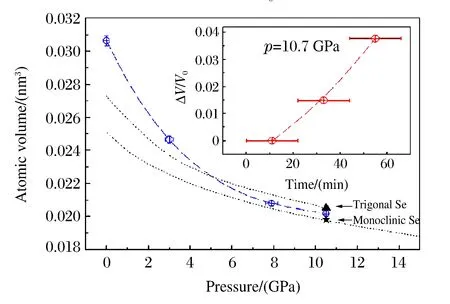
圖7 無定形Se的p-V曲線(插圖反映了無定形Se在結晶過程中的密度變化)[16]Fig.7 p-V curve of the amorphous Se (Theinsert shows how the density changes during the crystallization of amorphous Se)[16]
2014年Zeng等人[25]采用X射線成像方法探討了金屬玻璃的體積V與第一衍射峰位Q1的關系。他們用壓力作為密度增加的參數,通過XRD和顯微成像技術獲得相應的單胞尺度和體積,進而求出二者之間的冪次關系。為了獲得較大范圍的密度值,實驗中選取了體積模量很小的金屬玻璃(體積模量約41 GPa),加壓過程中金屬玻璃沒有發生結晶化及相變,而且加載過程幾乎是彈性壓縮,卸載后Q1回到了初始位置。為了準確測量高壓下樣品的密度,他們利用TXM技術(分辨率為30 nm)測量了高壓下樣品的體積,同時利用集成于巴黎-愛丁堡壓機(Paris-Edinburgh Cell)的聲速干涉儀和X射線投影成像方法,在低壓區對上述測量結果進行了驗證(見圖5)。結果表明,由Q1值得到的二者關系并非之前認為的各向同性的3次方關系,而是偏于分形結構的2.5次方關系,如圖6所示。2015年Lin等人[26]也采用上述方法對GeO2的高壓體積進行了測量。此外,Liu等人[16]根據XRD數據的精修結果,研究了無定形Sn在高壓結晶過程中的密度變化,發現Sn會發生約3.6%的密度變化。為了精確地原位測量無定形Se在結晶過程中的密度變化,Liu等人[16]在APS的2-BM線站上對無定形Se進行了高壓(壓力為10.7 GPa)X射線斷層掃描成像,如圖7所示,由于成像邊界存在模糊現象,在成像中通過上述方法轉換為密度測量。
對于高壓下液體和熔融態密度測量,早期使用沉降法,但是誤差較大,且實驗過程復雜。2011年Nishida等人[13]利用裝備在日本Spring-8同步輻射BL22XU線站上的DIA型壓機(采用圓柱形單晶藍寶石作為樣品容器),對液態硫化鐵進行了高溫高壓實驗,通過對y方向進行一維掃描,得到吸收曲線,進而擬合出樣品的密度,其中藍寶石的吸收系數和密度分別取自Chantler[27]的理論公式和Pavese[28]的EOS曲線。該方法所得密度與XRD所得固體密度在誤差范圍內相吻合。吸收曲線法的誤差來自于光子計數誤差、硫化鐵吸收系數的不確定性、擬合誤差以及多次測量的均方誤差,總誤差約為2%。2014年Chen等人[14]用氮化硼作為傳壓介質,通過樣品內紅寶石球的二維投影,擬合出硫化鐵液體的密度,所得結果與固體硫化鐵XRD結果的相對誤差為1%~2%。這種二維投影方法不局限樣品形狀,適用于樣品的高壓變形情況,而且避免了采用單晶藍寶石作為樣品腔時給密度測量帶來的額外影響,同時液體密度測量實驗設備簡單,光子計數誤差和擬合誤差降低。
3.2 地幔中鐵的輸運
地核的形成機制對于地球歷史的鑒定至關重要。以金屬地核(主要成分為鐵)為核心的地球圈層結構表明,地球中存在著一種機制,可以將鐵合金從硅酸鹽中分離,并運送至地核。地幔中鐵的分離和輸運過程是地球物理研究的重要內容。
雖然人們已對硅酸鹽顆粒間的熔融鐵合金做了大量的實驗研究,但是仍未完全理解地核的形成機制。這是因為之前的實驗研究是基于二維觀測結果,在研究三維熔融連通性時會出現一些誤判。同步輻射X射線斷層掃描能夠克服二維觀測的局限性,提供樣品的三維信息。 2011年Zhu等人[29]以不同熔體含量(質量分數)的橄欖-玄武巖作為樣品,研究了上地幔中熔融物的滲透性質,結果表明:當熔體含量為2%~20%時,熔體在顆粒邊緣形成連通的通道,并且高熔體含量(大于10%)樣品的熔融物還會大量分布在晶粒的邊界面上,而滲透率在此區間沒有明顯變化,表明孔隙率與熔體的拓撲分布無關,而滲透率依然可以利用熔體含量與滲透率的立方關系[30]進行近似。為了量化熔體網格的連通性,Zhu等人[29]將通道截面縮小到中心點,進行骨骼化算法分析,指出所有樣品中的連通網絡都是通過熔體的三重結點構成,與之前的經驗結果相同;但是由于X射線斷層掃描的分辨率為0.7 μm,對于熔體含量很小(2%)的情況,其連通性的參數測量會受到三維重建偽影的影響。
為了獲得更高的分辨率和襯度,需采用CDI技術對樣品進行成像。為了模擬地球上地幔的條件,Jiang等人[31]將圣卡羅橄欖石和鐵按質量比4∶1放入大壓機(Multi Anvil)內,在壓力為6 GPa、溫度為1 800 ℃的條件下經過1 h合成出樣品;在Spring-8同步輻射RIKEN光束線(BL29XUL)中,對合成的樣品等斜率(旋轉角為-69.4°~69.4°)地采集27幅圖像進行重建,所得電子密度形貌如圖8(a)所示。可見,3個不規則的顆粒互相連接(2.44 μm×2.31 μm×0.78 μm),顆粒的形狀、表面形貌和體積均與高溫高壓處理過程相關。根據質量m與電子個數Ne的關系m=2.05Ne,可以確定樣品的平均質量密度約為3.58 g/cm3,與此前的研究結果相吻合。為了研究富鐵相的二維分布,在上海同步輻射光源上進行TXM成像,在鐵吸收邊前、后得到了鐵的差圖像,將此結果與CDI重建的切片(16.3 nm厚,見圖8(b))進行比對,由CDI密度三維(3D)重建得到了富鐵相、FeS相和橄欖石相的質量密度區間,發現這些不同相的密度并非是預想的突變,而是連續變化;二維密度分布圖顯示,富鐵相和FeS相主要分布于顆粒的邊緣,另外還形成了一個鐵礦坑;三維形貌圖顯示,熔融鐵不僅形成孤立的球體(與之前的結果一致),還會形成不規則的三維形狀,從而更有利于鐵的輸運。當然這些熔融鐵的形成與局部的溫度和壓力、幾何結構以及三維滲透機制有關。

圖8 橄欖石-Fe-S樣品的相干衍射成像結果[31]Fig.8 Results of coherent diffraction imaging of olivine-Fe-S sample[31]
根據液態鐵在固體和各向同性基質中的滲透理論,當固-液界面能與固-固界面能的比值(二面角)變小時,熔融鐵從球狀變為凹面三棱鏡狀,從而有利于鐵的輸運,其中二面角可通過凹面三棱鏡之間的夾角進行測量。為了研究地核中鐵的輸運機制與高壓的關系,2013年Shi等人[32]將92%(質量分數)的頑輝石與8%的鐵-鎳合金混合,在常溫下加載至不同的壓力(25、39、52和64 GPa),然后利用雙面激光加熱至合金熔化而巖石仍為固態的最高溫度(由XRD表征狀態,分別為2 300、2 800、3 100和3 300 K)。由于固相具有各向異性,因此采用具有納米尺度的X射線透射顯微斷層掃描技術研究下地幔中熔融鐵的輸運與壓力的關系,其三維成像如圖9所示。從圖9可以看出,鐵由開始時孤立的Pocket(25和39 GPa)發展到形成連通網絡(52和64 GPa),巖石間隙的熔融鐵也從球形(25和39 GPa)轉變為枝叉形(52和64 GPa),對其二面角進行測量,得到最可幾二面角比值。當壓力較低時,二面角較大;隨著壓力的增加,二面角開始變小:表明壓力可使巖石中熔融鐵的滲透能力增強。分析64 GPa壓力下合成樣品的滲透性質,發現熔融鐵不僅分布在晶粒的邊緣,還擴散到晶粒的邊界面上。根據骨骼化算法,在該壓力點下,熔體主要通過三重和四重結點進行連通,與滲透分析結果一致,表明當壓力高于50 GPa時,固-液界面能降低到某臨界值以下,或者兩相結構發生變化,使巖石中的熔融鐵通過滲透進行連通。
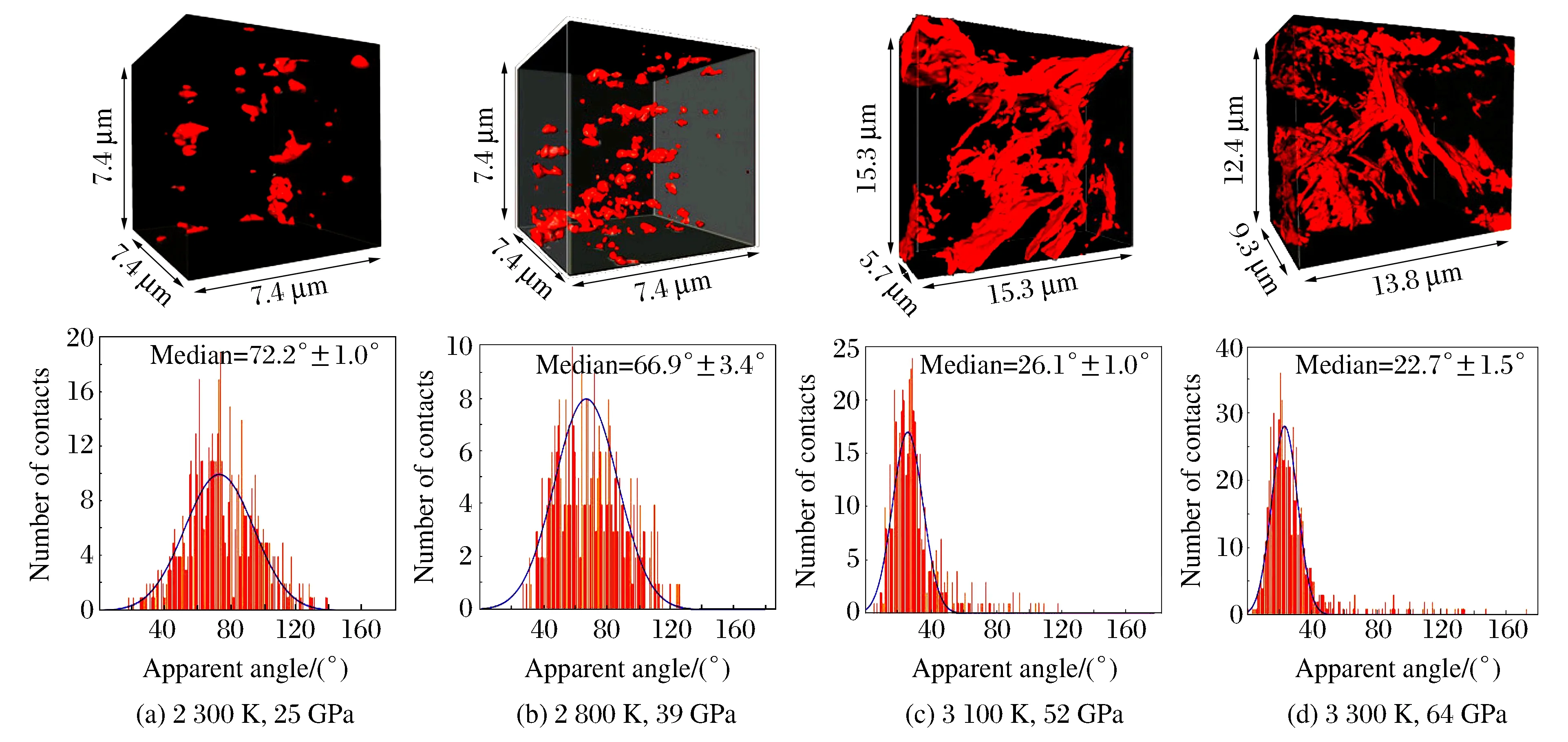
圖9 熔融鐵的三維空間分布和二面角統計分布[32]Fig.9 Three-dimensional spatial distribution of molten Fe and the statistical distribution of the dihedral angle[32]
3.3 相變演化及動力學過程
盡管XRD技術可用于研究原子排列變化所導致的結構相變,但相變核的分布和相變過程的可視化卻需要X射線成像技術完成。應用X射線成像技術可以得到密度、元素、價態等物理參數的分布,進而實時觀測相變的演化過程。
2004年,Katayama等人[33]在日本Spring-8同步輻射裝置的BL14B1線站上,利用X射線投影成像技術研究了磷在高溫高壓(六面頂壓機SMAP180)下的相變,通過X射線投影成像圖定性地觀察到樣品的相變過程(見圖10):在0.77 GPa、765 ℃條件下,樣品從初始的紅磷轉變為如圖10(a)所示的黑磷;在0.80 GPa、1 000 ℃條件下黑色區域變淡,由XRD峰可知此變化是黑磷相變為低密度流體態(Low Density Fluid Phase,LDFP);繼續加壓至0.86 GPa、溫度為995 ℃時,出現了一個球形的高密度區,此區域的衍射譜與LDFP明顯不同,表明新相出現;直至1.01 GPa時,此新相充滿整個樣品腔,XRD譜指出高密度液體相(High Density Liquid Phase,HDLP)的相變完成,從中可推測出0.86 GPa下的高密度球為HDLP相,結合HDLP相出現的影像可知,兩相的物理密度不同,界面張力的差別很大,一般條件下非極化分子液體的界面張力小于金屬液體,與之前電導率的理論和實驗結果相吻合[34-35]。

圖10 高溫高壓下磷的X射線投影成像圖[33]Fig.10 X-ray radiography imaging of P under high pressure and at high temperature[33]
對于某些材料,X射線成像技術還可以實現相變過程的五維(x,y,z,E,p)觀測,其中x、y、z為三維坐標,E為能量,p為壓力。BiNiO3材料在3~5 GPa時會由低壓相(Low Pressure Phase,LPP)變為高壓相(High Pressure Phase,HPP),同時伴隨著Ni元素電荷的轉移[36]。利用這一特點,2014年Liu等人[37]采用最近發展的X射線近邊吸收譜(X-Ray Absorption Near-Edge Spectrum,XANES)成像方法[38](TXM與XANES技術結合),將BiNiO3相變過程中的兩相分布隨壓力的變化呈現出來,圖11(a)顯示了3.89、4.17和4.55 GPa下HPP和LPP的三維分布圖。從圖11(a)中明顯看出,初始時由于壓力的不均勻造成HPP相分布不均勻,隨著壓力的增加,HPP相從幾個孤立的點出發,逐漸擴展并最后發展成連續的相體。另外,為深入研究相變過程中相邊界的結構,從某個二維切片(見圖11(b))入手,根據Canny邊緣探測算法,計算出3個壓力點下的相邊界(見圖11(c)),以分析相變時相邊界的演化情況。圖11(c)中,邊界突出部分的相變速度低,凹下部分的相變速度高,最終達到類似球形的狀態,如同熔融鐵的滲透過程。正是這些不規則的相邊界驅動著樣品由LPP相變為高壓下相對穩定的HPP。通過五維探測可以實現相變過程中不同相的體積和邊界形狀的測量,洞察相變過程的潛在機制。

圖11 不同壓力下LPP(紅色)和HPP(綠色)的BiNiO3粒子的三維分布(a)二維切片(b) 和二維相界(c)[38]Fig.11 Three-dimensional spatial distributions (a),slices (b),and phase boundaries (c) of LPP (red) and HPP (green) of BiNiO3 particles under different pressures[38]
對于玻璃相變,目前還沒有基本理論可以描述。2014年Li等人[39]采用顆粒狀粒子混合液體模型研究了玻璃或膠體內的動力學作用機制:通過機械振蕩粒子,利用X射線快速斷層掃描技術,獲得了不同時間(震蕩次數)下粒子的分布,進而得到聚合配位數和局部結構等信息;根據聚合配位數隨著時間變化的曲線,推測出成鍵是推動聚合的主要動力;同時,根據模型結構趨于穩定時局部區域存在著大量的五次對稱結構,確定出玻璃相變的動態結構作用機理應為多面體短程有序[40],并非晶格有序[41]。利用X射線成像技術研究基于玻璃相變的粒子模型,也是目前非晶動力學研究的一大熱點。
3.4 其他應用
X射線成像技術還可以用于黏度[42-44]、聲速[45]、應力-應變分布[46-47]的測量。除了以上介紹的X射線成像技術之外,還有很多有潛力的成像技術能在高壓領域中運用,如X射線光子相關譜、超快X射線相干衍射等。X射線光子相關譜是通過不同時刻相干衍射圖的互相關,研究材料內磁疇(約1 μm)隨時間的變化[48],因此也可以利用不同壓力下相干衍射圖的互相關,研究磁疇隨壓力的變化。超快X射線相干衍射是通過調節泵浦激光和相干X光的時間差,得到樣品泵浦后不同時刻的三維相位以及三維聲子的剪切振動[49],如果加入高壓維度,便可獲得壓力與聲子剪切振動的關系。
4 總結與展望
X射線成像技術與高壓的結合離不開高壓加載技術和三維圖像重建方法的不斷突破。三維成像需要多角度下樣品的投影信息:對于大壓機,其固定環需采用鋁合金或其他透光材質[18];對于傳統的金剛石壓機,其透光方向十分有限,因此先后出現了全景壓機[50-51]、十字型壓機[25],甚至一字型壓機[52]。全景壓機有3個窗口,每個窗口沿赤道方向和經度方向的開口角分別為105°和68°,可達到153 GPa的壓力[50]。十字型壓機的外形類似于十字架,有4個窗口,X光被螺絲阻擋的部分只有30°[25],但是目前所能達到的最高壓力有限。盡管如此,仍然存在投影死角區,針對這一問題,人們提出了多種三維圖像重建方法。2008年Liu等人[16]采用小角度間隔的等角掃描,重建出樣品后,計算得到平均密度;2012年Wang等人[19]提出利用代數迭代方法逐漸逼近真實的三維分布;采用應用于電子斷層重建和相干衍射有限角度掃描重建的等斜率斷層掃描(Equally Sloped Tomography)方法[53],可將投影數量減小60%~70%[54],對于高壓X射線成像技術的應用具有重要意義。此外,為了能在更高的壓力下應用X射線成像技術,通過設計大壓機中的壓砧角度、尖端大小、固定環尺寸,以及金剛石壓機中的封墊組合,以期達到更高的壓力[18]。
X射線成像技術在高壓下的進一步應用仍有待于其技術本身的發展,例如:提高成像的空間分辨率和襯度分辨率,從而更精準地呈現樣品的三維結構分布,進而獲得相關的物理參數。X射線成像技術與其他技術相結合可以獲得更豐富的信息,也是X射線成像技術應用的發展趨勢,比如:將X射線吸收與掃描方式相結合,可進行密度測量;在多晶X射線勞埃衍射實驗中運用針孔掃描技術,可測量變形晶體中的應變分布;將TXM與XANES技術相結合,可得到不同價態的三維分布。
[1] THURING T,ABIS M,WANG Z,et al.X-ray phase-contrast imaging at 100 keV on a conventional source [J].Sci Rep,2014,1(1):1-4.
[2] WILKINS W S,GUREYEV T E,GAO D,et al.Phase-contrast imaging using polychromatic hard X-rays [J].Nature,1996,384(28):335-338.
[3] WANG Y,YUN W,JACOBSEN C.Achromatic Fresnel optics for wideband extreme-ultraviolet and X-ray imaging [J].Nature,2003,424(6944):50-53.
[4] MIAO J,CHARALAMBOUS P,KIRZ J,et al.Extending the methodology of X-ray crystallography to allow imaging of micrometre-sized non-crystalline specimens [J].Nature,1999,400(6742):342-344.
[5] TIAN Y,XU B,YU D,et al.Ultrahard nanotwinned cubic boron nitride [J].Nature,2013,493(7432):385-388.
[6] OVSYANNIKOV S,SHCHENNIKOV V.Pressure-tuned colossal improvement of thermoelectric efficiency of PbTe [J].Appl Phys Lett,2007,90(12):122103.
[7] MURAKAMI M,HIROSE K,KAWAMURA K,et al.Post-perovskite phase transition in MgSiO3[J].Science,2004,304(5672):855-858.
[8] ZHANG L,MENG Y,YANG W,et al.Disproportionation of (Mg,Fe)SiO3perovskite in Earth’s deep lower mantle [J].Science,2014,344(6186):877-882.
[9] CHAPMAN H N,BARTY A,MARCHESINI S,et al.High-resolutionabinitiothree-dimensional X-ray diffraction microscopy [J].J Opt Soc Am A,2006,23(5):1179-1200.
[10] KATAYAMA Y.Density measurements of non-crystalline materials under high pressure and high temperature [J].High Pressure Res,1996,14(4/5/6):383-391.
[11] KATAYAMA Y,TSUJI K,CHEN J Q,et al.Density of liquid tellurium under high pressure [J].J Non-Cryst Solids,1993,156/157/158:698-690.
[12] SANLOUP C,GUYOT F,GILLET P,et al.Density measurements of liquid Fe-S alloys at high-pressure [J].Geophys Res Lett,2000,27(6):811-814.
[13] NISHIDA K,OHTANI E,URAKAWA S,et al.Density measurement of liquid FeS at high pressures using synchrotron X-ray absorption [J].Am Mineral,2011,96(5/6):864-868.
[14] CHEN J,YU T,HUANG S,et al.Compressibility of liquid FeS measured using X-ray radiograph imaging [J].Phys Earth Planet Inter,2014,228:294-299.
[15] HONG X,SHEN G,PRAKAPENKA V B,et al.Density measurements of noncrystalline materials at high pressure with diamond anvil cell [J].Rev Sci Instrum,2007,78(10):103905.
[16] LIU H,WANG L,XIAO X,et al.Anomalous high-pressure behavior of amorphous selenium from synchrotron X-ray diffraction and microtomography [J].Proc Natl Acad Sci,2008,105(36):13229-13234.
[17] XIAO X,LIU H,WANG L,et al.Density measurement of samples under high pressure using synchrotron microtomography and diamond anvil cell techniques [J].J Synchrotron Rad,2010,17(3):360-366.
[18] WANG Y,UCHIDA T,WESTFERRO F,et al.High-pressure X-ray tomography microscope:synchrotron computed microtomography at high pressure and temperature [J].Rev Sci Instrum,2005,76(7):073709.
[19] WANG J,YANG W,WANG S,et al.High pressure nano-tomography using an iterative method [J].J Appl Phys,2012,111(11):112626.
[20] YAVARI A R,MOULEC A L,INOUE A,et al.Excess free volume in metallic glasses measured by X-ray diffraction [J].Acta Mater,2005,53(6):1611-1619.
[21] CADIEN A,HU Q Y,MENG Y Q,et al.First-order liquid-liquid phase transition in cerium [J].Phys Rev Lett,2013,110(12):105503.
[22] MIRACLE D B.A structural model for metallic glasses [J].Nat Mater,2004,3(10):697-702.
[23] CHENG Y Q,MA E,SHENG H W.Atomic level structure in multicomponent bulk metallic glass [J].Phys Rev Lett,2009,102(24):245501.
[24] MEADE C,HEMLEY R J,MAO H K.High-pressure X-ray diffraction of SiO2glass [J].Phys Rev lett,1992,69(9):1387-1390.
[25] ZENG Q,KONO Y,LIN Y,et al.Universal fractional noncubic power law for density of metallic glasses [J].Phys Rev Lett,2014,112(18):185502.
[26] LIN Y,ZENG Q,YANG W,et al.Pressure-induced densification in GeO2glass:a transmission X-ray microscopy study [J].Appl Phys Lett,2013,103(26):261909.
[27] CHANTLER C T.Theoretical form factor,attenuation,and scattering tabulation forZ=1-92 fromE=1-10 eV toE=0.4-1.0 MeV [J].J Phys Chem Ref Data,1995,24(1):71-643.
[28] PAVESE A.Pressure-volume-temperature equations of state:a comparative study based on numerical simulations [J].Phys Chem Miner,2002,29(1):43-51.
[29] ZHU W,GAETANI G A,FUSSEIS F,et al.Microtomography of partially molten rocks:three-dimensional melt distribution in mantle peridotite [J].Science,2011,332(6025):88-91.
[30] WARK D A,WATSON E B.Grain-scale permeabilities of texturally equilibrated,monomineralic rocks [J].Earth Planet Sci Lett,1998,164(3/4):591-605.
[31] JIANG H,XU R,CHEN C,et al.Three-dimensional coherent X-ray diffraction imaging of molten iron in mantle olivine at nanoscale resolution [J].Phys Rev Lett,2013,110(20):205501.
[32] SHI C Y,ZHANG L,YANG W,et al.Formation of an interconnected network of iron melt at Earth’s lower mantle conditions [J].Nat Geosci,2013,6(11):971-975.
[33] KATAYAMA Y,INAMURA Y,MIZUTANI T,et al.Macroscopic separation of dense fluid phase and liquid phase of phosphorus [J].Science,2004,306(5697):848-851.
[34] SENDA Y,SHIMOJO F,HOSHINO K.The metal-nonmetal transition of liquid phosphorus byabinitiomolecular-dynamics simulations [J].J Phys Condens Matter,2002,14(14):3715-3723.
[35] HOHL D,JONES R O.Polymerization in liquid phosphorus:simulation of a phase transition [J].Phys Rev B,1994,50(23):17047.
[36] AZUMA M,CHEN W,SEKI H,et al.Colossal negative thermal expansion in BiNiO3induced by intermetallic charge transfer [J].Nat Commun,2011,2(6):347-351.
[37] LIU Y,WANG J,AZUMA M,et al.Five-dimensional visualization of phase transition in BiNiO3under high pressure [J].Appl Phys Lett,2014,104(4):043108.
[38] MEIRER F,CABANA J,LIU Y,et al.Three-dimensional imaging of chemical phase transformations at the nanoscale with full-field transmission X-ray microscopy [J].J Synchrotron Rad,2011,18(5):773-781.
[39] LI J,CAO Y,XIA C,et al.Similarity of wet granular packing to gels [J].Nat Commun,2014,5(9):1-7.
[40] ZACCARELLI E,LU P J,CIULLA F,et al.Gelation as arrested phase separation in short-ranged attractive colloid-polymer mixtures [J].J Phys Condens Matter,2008,20(49):494242.
[41] ROYALL C P,WILLIAMS S R,OHTSUKA T,et al.Direct observation of a local structural mechanism for dynamic arrest [J].Nat Mater,2008,7(7):556-561.
[42] URAKAWA S,TERASAKI H,FUNAKOSHI K,et al.Radiographic study on the viscosity of the Fe-FeS melts at the pressure of 5 to 7 GPa [J].Am Mineral,2001,86(4):578-582.
[43] KONO Y,PARK C,KENNEY-BENSON C,et al.Toward comprehensive studies of liquids at high pressures and high temperatures:combined structure,elastic wave velocity,and viscosity measurements in the Paris-Edinburgh cell [J].Phys Earth Planet Inter,2014,228:269-280.
[44] FUNAKOSHI K,NOZAWA A.Development of a method for measuring the density of liquid sulfur at high pressures using the falling-sphere technique [J].Rev Sci Instrum,2012,83(10):103908.
[45] LI B,KUNG J,LIEBERMANN R C.Modern techniques in measuring elasticity of Earth materials at high pressure and high temperature using ultrasonic interferometry in conjunction with synchrotron X-radiation in multi-anvil apparatus [J].Phys Earth Planet Inter,2004,143/144:559-574.
[46] LARSON B C,YANG W,ICE G E,et al.Three-dimensional X-ray structural microscopy with submicrometre resolution [J].Nature,2002,415(6874):887-890.
[47] YANG W,HUANG X,HARDER R,et al.Coherent diffraction imaging of nanoscale strain evolution in a single crystal under high pressure [J].Nat Commun,2009,8(4):291-298.
[48] SHPYRKO O G,ISAACS E D,LOGAN J M,et al.Direct measurement of antiferromagnetic domain fluctuations [J].Nature,2007,447(3):68-71.
[49] TRIGO M,FUCHS M,CHEN J,et al.Fourier-transform inelastic X-ray scattering from time- and momentum-dependent phonon-phonon correlations [J].Nat Phys,2013,9(12):790-794.
[50] XU J A,MAO H K,HEMLEY R J,et al.The moissanite anvil cell:a new tool for high-pressure research [J].J Phys Condens Matter,2002,14(44):11543-11548.
[51] MAO H K,XU J,STRUZHKIN V V,et al.Phonon density of states of iron up to 153 gigapascals [J].Science,2001,292(5518):914-916.
[52] URAKAWA S,TERASAKI H P,FUNAKOSHI K,et al.Development of high pressure apparatus for X-ray microtomography at Spring-8 [J].J Phys Conf Ser,2010,215(1):012026.
[53] MIAO J,FORSTER F,LEVI O.Equally sloped tomography with oversampling reconstruction [J].Phys Rev B,2005,72(5):052103.
[54] FAHIMIAN B,MAO Y,CLOETENS P,et al.Low-dose X-ray phase-contrast and absorption CT using equally sloped tomography [J].Phys Med Biol,2010,55(18):5383-5400.
猜你喜歡
小學科學(學生版)(2021年5期)2021-07-22 02:40:06
中學生數理化·八年級物理人教版(2019年9期)2019-11-25 07:33:02
中學生數理化·八年級物理人教版(2019年3期)2019-04-25 06:20:54
中學生數理化·八年級物理人教版(2018年3期)2018-05-31 08:52:45
數學小靈通(1-2年級)(2017年10期)2017-11-08 08:39:45
軍事文摘·科學少年(2017年4期)2017-06-20 23:25:16
軍事文摘·科學少年(2017年2期)2017-04-26 21:58:43
中學生數理化·八年級物理人教版(2016年3期)2016-04-07 04:49:32
少兒科學周刊·兒童版(2016年1期)2016-03-14 03:52:21
閱讀與作文(小學低年級版)(2015年4期)2015-04-29 00:00:00
