6例Gitelman綜合征患者的臨床及基因學分析
2016-07-22 02:44:06苗苗趙超群劉靜李金慧王曉黎單忠艷
中國醫科大學學報 2016年7期
關鍵詞:基因突變
苗苗,趙超群,劉靜,李金慧,王曉黎,單忠艷
(中國醫科大學附屬第一醫院內分泌科,內分泌研究所,遼寧省內分泌疾病重點實驗室,沈陽 110001)
?
6例Gitelman綜合征患者的臨床及基因學分析
苗苗,趙超群,劉靜,李金慧,王曉黎,單忠艷
(中國醫科大學附屬第一醫院內分泌科,內分泌研究所,遼寧省內分泌疾病重點實驗室,沈陽 110001)
摘要在低鉀血癥患者中篩選出6例臨床診斷符合Gitelman綜合征(GS)的患者,分析其臨床資料,對符合臨床診斷患者的致病基因SLC12A3及經典型Bartter綜合征(BS)的致病基因CLCNKB進行直接測序,尋找致病突變位點,探討基因型與表型的聯系。基因測序發現其中3例患者存在SLC12A3的致病突變。臨床疑診GS的患者需通過基因診斷確證,不攜帶基因變異的患者可能存在其他原因導致相似臨床表型。
關鍵詞Gitelman綜合征;SLC12A3基因;CLCNKB基因;基因突變
網絡出版地址
Gitelman綜合征(Gitelman syndrome,GS)是一種常染色體隱性遺傳的腎小管疾病,其臨床表現與Bartter綜合征(Bartter syndrome,BS)相似,主要臨床特征為低鉀血癥、低鎂血癥、低尿鈣、醛固酮水平增高但血壓正常。GS在高加索人群中患病率大約為1/40 000,雜合子攜帶率高達1%,為最常見的遺傳性腎小管疾病之一[1]。目前尚無對中國人GS發病率的數據統計。GS與位于16號染色體長臂上的SLC12A3基因突變有關[2]。SLC12A3基因編碼噻嗪類利尿劑敏感的Na+/Cl-共同轉運體(thiazide sensi?tive sodium chloride cotransporter,Na?Cl cotransport?er,NCCT),目前已發現超過140種SLC12A3基因的突變類型[3]。在少數的GS患者中發現編碼氯通道的CLCNKB基因突變。
本研究對臨床診斷為GS患者的病史、臨床表現、實驗室和影像學檢查進行分析,并對其致病靶基因進行測序,探討基因型與表型的聯系。
1 材料與方法
1.1研究對象
選擇2014年至2015年于中國醫科大學第一附屬醫院內分泌科住院的低鉀血癥患者30例,根據GS的臨床診斷標準[4]篩選出符合診斷標準的患者6例,其中男4例,女2例,年齡18~65歲,病程1~10年。
1.2診斷標準
低鉀血癥、堿中毒、高尿鉀(>25 mmol/24 h)、低血鎂(<0.66 mmol/L)、低尿鈣肌酐比(<0.2),腎素血管緊張素活性升高但血壓正常,且排除轉移性低鉀、胃腸道失鉀、腎小管酸中毒,以及緩泄劑、利尿劑或乙醇的用藥史[4]。
1.3病例簡介
以下病例均排除其他低鉀常見病因。
病例1:18歲男性,乏力等癥狀不明顯,十余年來反復出現低血鉀,經補鉀對癥治療后低血鉀仍未糾正。
病例2:22歲男性,四肢麻木、乏力、易摔倒2年,于醫院就診發現低鉀,經對癥補鉀治療后上述情況仍反復出現。
病例3:44歲男性,10余年發作性下肢無力病史,嚴重時蹲起困難,病情逐漸發展至頻繁發作性肢體軟癱。
病例4:28歲女性,乏力、心悸病史5年,每次發病經對癥治療后好轉,此次發病血鉀最低達1.88 mmol/L,臨床特點為發病年齡早,血壓偏低。
病例5:55歲男性,下肢乏力及麻木病史10余年,發現血鉀低9年,經對癥補鉀后仍不易糾正。臨床特點為患者發病年齡較晚,同時伴有糖尿病及糖尿病周圍神經病變。
病例6:65歲女性,周身乏力、氣短伴惡心癥狀半年,化驗發現血鉀2.4 mmol/L,每次發作對癥補鉀后癥狀好轉,臨床特點為發病年齡晚,血壓正常。
1.4檢測方法
血電解質以全自動生化儀測定。促腎上腺皮質激素(adreno cortico tropic hormone,ACTH)、皮質醇采用免疫化學發光法測定,血腎素、血管緊張素、血醛固酮采用放射免疫法測定。
1.5基因檢測
使用DNA提取試劑盒(天根生化科技北京有限公司)提取外周血基因組DNA,對PCR產物純化(天根生化科技北京有限公司瓊脂糖凝膠回收試劑盒),再對SLC12A3基因及CLCNKB基因進行全部外顯子以及外顯子-內含子交接部分的直接測序(上海新培晶檢驗所及北京六合華大基因科技股份有限公司),測序引物見以往文獻[5]。
1.6應用在線軟件預測突變的功能
應用在線蛋白質預測軟件PolyPhen?2(http:// genetics.bwh.harvard.edu/pph2/)和SIFT(http://prove?an.jcvi.org/)對發現的突變位點進行功能預測。
2 結果
6例患者均存在高尿鉀,低鎂血癥、低尿鈣,腎素血管緊張素活性升高但血壓正常,在排除轉移性低鉀、胃腸道失鉀、腎小管酸中毒,以及致鉀流失的用藥史之后臨床診斷GS,見表1。
基因診斷結果(表2,圖1)提示3例患者存在導致SLC12A3基因功能減低的突變(L849H,T60M和L671P)。應用在線蛋白質功能預測軟件PolyPhen?2和SIFT對此3處突變進行預測提示均會導致蛋白質的功能減低,見表3。
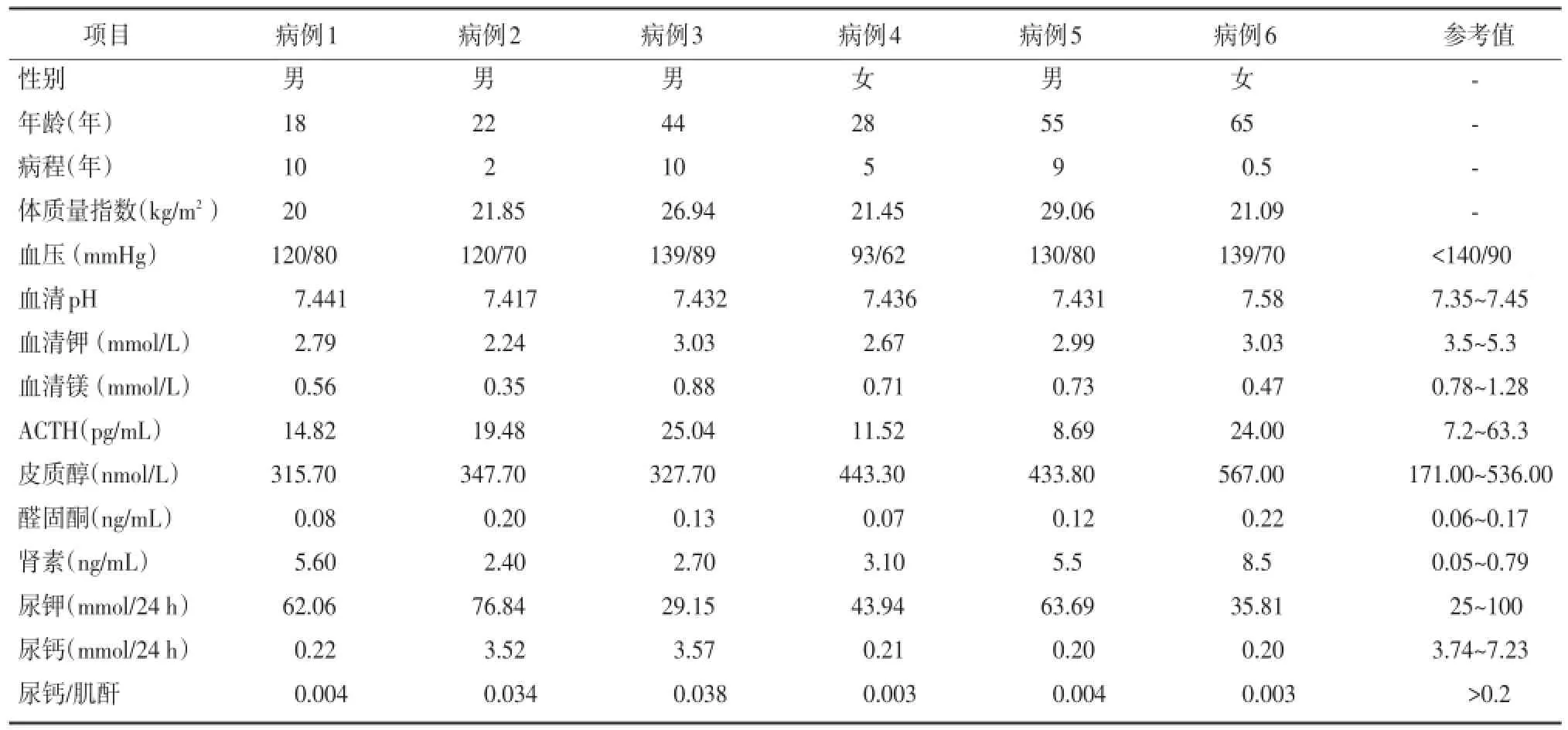
表1 病例基本資料及實驗室檢查結果

表2 6例基因檢測結果
3 討論
Gitelman[6]在1966年首次報道了3例22~47歲的女性患者,臨床表現類似BS,但同時伴有低血鎂、低尿鈣,后被稱為GS[7]。GS發病與位于16號染色體長臂上的SLC12A3基因突變有關[8-9],目前已發現140多個SLC12A3基因突變位點(人類基因突變數據庫HGMD),包括錯義突變、剪切突變、無義突變、讀碼框位移突變等,其中大部分以錯義突變為主,復合雜合突變多于純合子突變,無熱點突變發現。國內對一組臨床診斷為GS的患者進行基因學檢測,結果發現T60M可能是中國患病人群最常見的突變位點[10],但并非所有的GS患者均攜帶SLC12A3基因突變,提示可能有其他遺傳位點表現為相似的表型[11]。
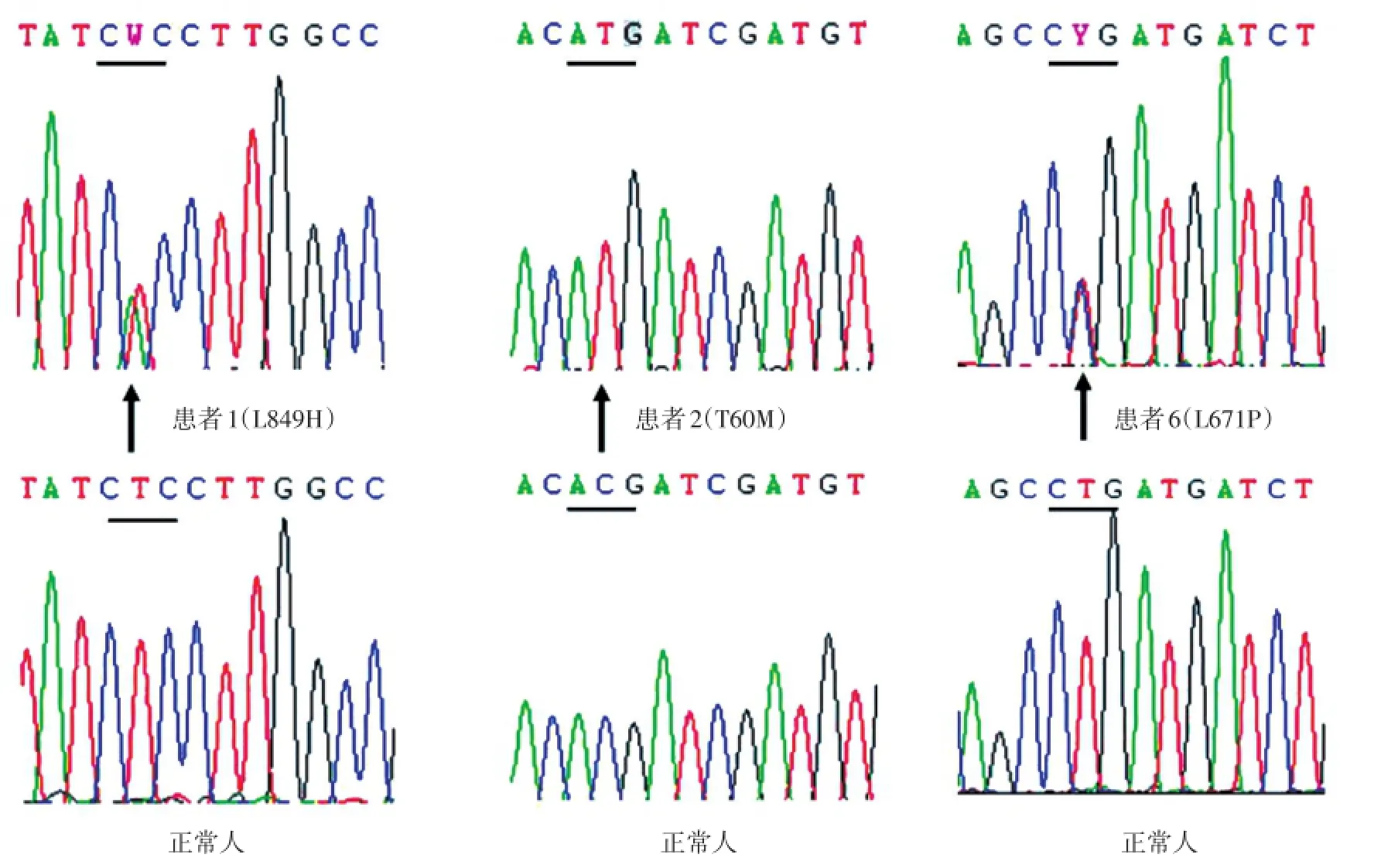
圖1 SLC12A3基因檢測結果

表3 SLC12A3基因突變預測結果
本研究中6例患者均為近期于我科就診的低鉀血癥患者,共同特點是存在低鉀血癥、低鎂血癥、腎性失鉀但血壓正常。因為BS與GS的臨床表型部分重疊,因此本研究通過進一步對靶基因SLC12A3和CLCNKB的檢測來明確是否存在導致GS的遺傳學基礎。3例患者發現了SLC12A3基因突變,其中患者2的突變類型為純合型T60M,曾有報道[10]認為是中國人中GS患者特有的常見突變類型。患者1的突變類型為雜合型L849H,曾有文獻[12-14]報道在日本GS患者中有相同基因型,并認為其僅存在于日本人種中。筆者認為L849H雜合突變不足以導致患者出現GS的臨床表型,考慮可能存在基因調節區域的變異或者其他導致相似表型的基因中存在未知的變異。本研究中僅對導致經典型BS的靶基因進行了分析,未發現致病位點。另外,在日本人中的研究[15]表明攜帶L849H雜合突變的個體血鉀與正常人相比差異具有統計學意義,甚至提出了“攜帶SLC12A3基因功能性雜合突變是影響血壓水平的一個重要因素”的觀點[16]。本研究中患者6的突變類型為雜合型L671P突變,目前尚未見國內外的文獻報道,通過在線蛋白質功能預測軟件預測,與T60M 和L849H同樣位于蛋白質保守區域的L671P突變會損害蛋白質的正常功能,考慮可能為一處新的導致GS表型的基因突變類型。
此6例患者確診后均接受補鉀聯合應用門冬氨酸鉀鎂和(或)安體舒通等治療,低鉀的臨床癥狀(乏力、軟癱等)均得到緩解,但尚未完全糾正低血鎂、低血鉀,說明GS的離子紊亂不易糾正,需要多種藥物聯合長期應用。
GS是一種預后良好、進展緩慢的疾病,迄今報道的發展到終末期尿毒癥的GS患者僅有2例[17-18]。但GS長期發展會影響患者生活質量,并有引起慢性腎功能不全的危險。基因診斷的意義在于早期確診后進行針對性的治療,本病尚無法根治,治療應對癥補鉀、補鎂,前列腺素合成酶抑制劑、醛固酮拮抗劑等多種藥物聯合應用。BS患者的前列腺素E2往往是升高的,而GS患者絕大多數前列腺素E2為正常,據此有研究[19]認為應用環氧化酶抑制劑治療GS效果不佳。本研究中的6例病例提示臨床工作中應注意頑固性低鉀血癥的患者是否存在GS,對高度懷疑GS的患者應盡早行基因診斷檢測,從而提高對GS的認識和診治水平,提高患者生活質量。
參考文獻:
[1]KNOERS NV,LEVTCHENKO EN.Gitleman syndrom[J].Or?phanet J Rare Dis,2008,3:22.
[2]GALLI?TSINOPOULOU A,PATSEADOU M,HATZIDIMITRIOU A.Gitelman syndrome:first report of genetically established diagno?sis in Greece[J].Hippokratia,2010,14(1):42-44.
[3]SIMON DB,NELSON WC,BIA MJ.Gitelman variant of Bartter's syndrome,inherited hypokalaemic alkalosis,is caused by mutations in the thiazide?sensitive Na?Cl cotransporter[J].Nat Genet,1996,12(1):24-30.
[4]SINHA A,LNěNIKA P,BASU B.Gitelman syndrome:novel muta?tion and long?term follow?up[J].J Clin Endocrinol Metab,2012,16 (2):306-309.
[5]MONNENS L,BINDELS R,GRUNFELD JP.Gitelman syndrome comes of age[J].Nephro1 Dial Transplant,1998,13(7):1617-1619.
[6]楊國慶,趙蕾,席文琪,等.Gitelman綜合征9例臨床分析[J].中華內科雜志,2006,45(8):650-653.
[7]LEMMINK HH,KNOERS NV,KáROLYI L.Novel mutations in the thiazide?sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C?terminal domain [J].Kidney Int,1998,54(3):720-730.
[8]GITELMAN HJ,GRAHAM JB,WELT LG.A new familial disorder characterized by hypokalemia and hypomagnesemia[J].Trans As?soc Am Physicians,1966,79(3):221-235.
[9]GALLI?TSINOPOULOU A,PATSEADOU M,HATZIDI MITRIOU A.Gitelman syndrome:first report of genetically established diagno?sis in Greece[J].Hippokratia,2010,14(1):42.
[10]邵樂平,任紅,王偉銘.Gitelman綜合征SLC12A3基因突變研究[J].中華腎臟病雜志,2007,66(23):351-356.
[11]QIN L,SHAO L,REN H.Identification of five novel variants in the thiazide?sensitive NaCl co?transporter gene in Chinese pa?tients with Gitelman syndrome[J].Nephrology(Carlton),2009,14(1):52-58.
[12]MONKAWA T,KURIHARA I,KOBAYASHI K.Novel mutations in thiazide?sensitive Na?Cl cotransporter gene of patients with Gitelman's syndrome[J].J Am Nephrol,2000,11(1):65-70.
[13]MAKI N,KOMATSUDA A,WAKUI H.Four novel mutations in the thiazide?sensitive Na?Cl co?transporter gene in Japanese pa?tients with Gitelman's syndrome[J].Nephrol Dial Transplant,2004,19(7):1761-1766.
[14]AOI N,NAKAYAMA T,TAHIRA Y,et al.Two novel genotypes of the thiazide?sensitive Na?Cl cotransporter(SLC12A3)gene in pa?tients with Gitelman's syndrome[J].Endocrine,2007,31(2):149-153.
[15]NARABA H,KOKUBO Y,TOMOIKE H,et al.Functional confir?mation of Gitelman's Syndrome mutations in Japanese[J].Hyper?tens Res,2005,28(10):805-809.
[16]TAGO N,KOKUBO Y,INAMOTO N,et al.A high prevalence of Gitelman syndrome mutations in Japanese[J].Hypertens Res,2004,27(5):327-331.
[17]BONFANTE L,DAVIS PA,SPINELLO M,et al.Chronic renal fail?ure,end?stage renal disease,and peritoneal dialysis in Gitelman syndrome[J].Am J Kidney Dis,2001,38(1):165-168.
[18]CALò LA,MARCHINI F,DAVIS PA,et al.Kidney transplant in Gitelman's syndrome:report of the first case[J].Nephrol,2003,16(1):144-147.
[19]KURTZ I.Molecular pathogenesis of Bartter's and Gitelman[J]. Kidney Int,1998,54(4):1396-1410.
(編輯于溪)
網絡出版時間:
中圖分類號R589.9
文獻標志碼A
文章編號0258-4646(2016)07-0649-04
DOI:10.12007/j.issn.0258?4646.2016.07.016
基金項目:國家自然基金青年基金(81200653);衛生部國家臨床重點專科資助
作者簡介:苗苗(1989-),女,碩士研究生.
通信作者:王曉黎,E-mail:wlittlepear@163.com
收稿日期:2015-10-23
AClinicalandGeneticAnalysisof6CasesofGitelmanSyndrome
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
