常染色體顯性遺傳骨硬化癥Ⅱ型氯離子第七通道蛋白基因突變1例檢測
2016-08-11 00:35:25龐倩倩
中西醫結合心腦血管病雜志 2016年13期
龐倩倩,董 進
?
常染色體顯性遺傳骨硬化癥Ⅱ型氯離子第七通道蛋白基因突變1例檢測
龐倩倩,董進
山西醫科大學第一醫院(太原 030001)
關鍵詞:遺傳性骨病;骨硬化癥;基因突變;氯離子第七通道蛋白;常染色體;檢測
骨硬化癥是一類以骨密度增高,破骨細胞吸收功能障礙為主要特點的遺傳性骨病,根據其臨床表現和致病基因的不同可分為常染色體顯性遺傳骨硬化癥(autosomal dominant inheritance osteopetrosisADO),常染色體隱性遺傳骨硬化癥(autosomal recessive inheritance osteopetrosis,ARO)和罕見的X染色體連鎖隱性遺傳骨硬化癥(X-linked osteopetrosis,XLO)。骨硬化癥的臨床表現具有廣泛的異質性,如貧血、全血細胞減少、膿毒血癥、繼發性的肝脾腫大等。然而部分病人亦可無癥狀或癥狀較輕微,僅僅通過骨骼的影像學檢查才可以發現。在影像學上該疾病通常表現為“中心性骨硬化”和“彌漫性骨硬化”兩種類型,而這兩種類型均由于骨脆性增加而引起的骨折風險率增加[1]。根據其臨床表現不同,ADO又可分為:常染色體顯性遺傳骨硬化癥良性Ⅰ型(ADO-Ⅰ),良性Ⅱ型(ADO-Ⅱ),良性Ⅲ型(ADO-Ⅲ)[1-2],其中ADO-Ⅱ最為常見。該型具有典型的ADO影像學特征,主要的并發癥體現在骨骼系統,包括病理性骨折,脊柱側凸等[3]。ADO-Ⅱ主要是由位于染色體16p13,編碼氯離子第7通道蛋白(CLCN7)基因突變引起的[4]。目前國內罕有關于該疾病基因突變的報道,且尚未建立中國人群關于該疾病的基因突變譜,因此對骨硬化病人進行基因分析具有很重要的臨床意義。
本研究擬通過對1例常染色體顯性遺傳的骨硬化癥家系進行臨床及基因學特征分析,探討該疾病的致病機制。
1資料與方法
1.1研究對象先證者,男性,18歲。2015年因“骨硬化癥”,就診于我科門診。病人第一胎,足月順產,出生體重、身長、出牙時間均不詳。智力未見落后。實驗室檢查結果:生化指標未見明顯異常,全血細胞計數,血鈣,血磷,血堿性磷酸酶,血肌酐,血iPTH水平均處于正常范圍;影像學特征:頭顱正側位片示:顱骨骨皮質密度增厚,板障增厚,顱底骨質增厚硬化;胸腰椎正側位相示:椎體上下終板骨密度增厚,中央密度相對較低,呈“三明治”樣改變(見圖1)。家族史中未有類似癥狀。病人及其父母均被告知研究目的,并簽署知情同意書。
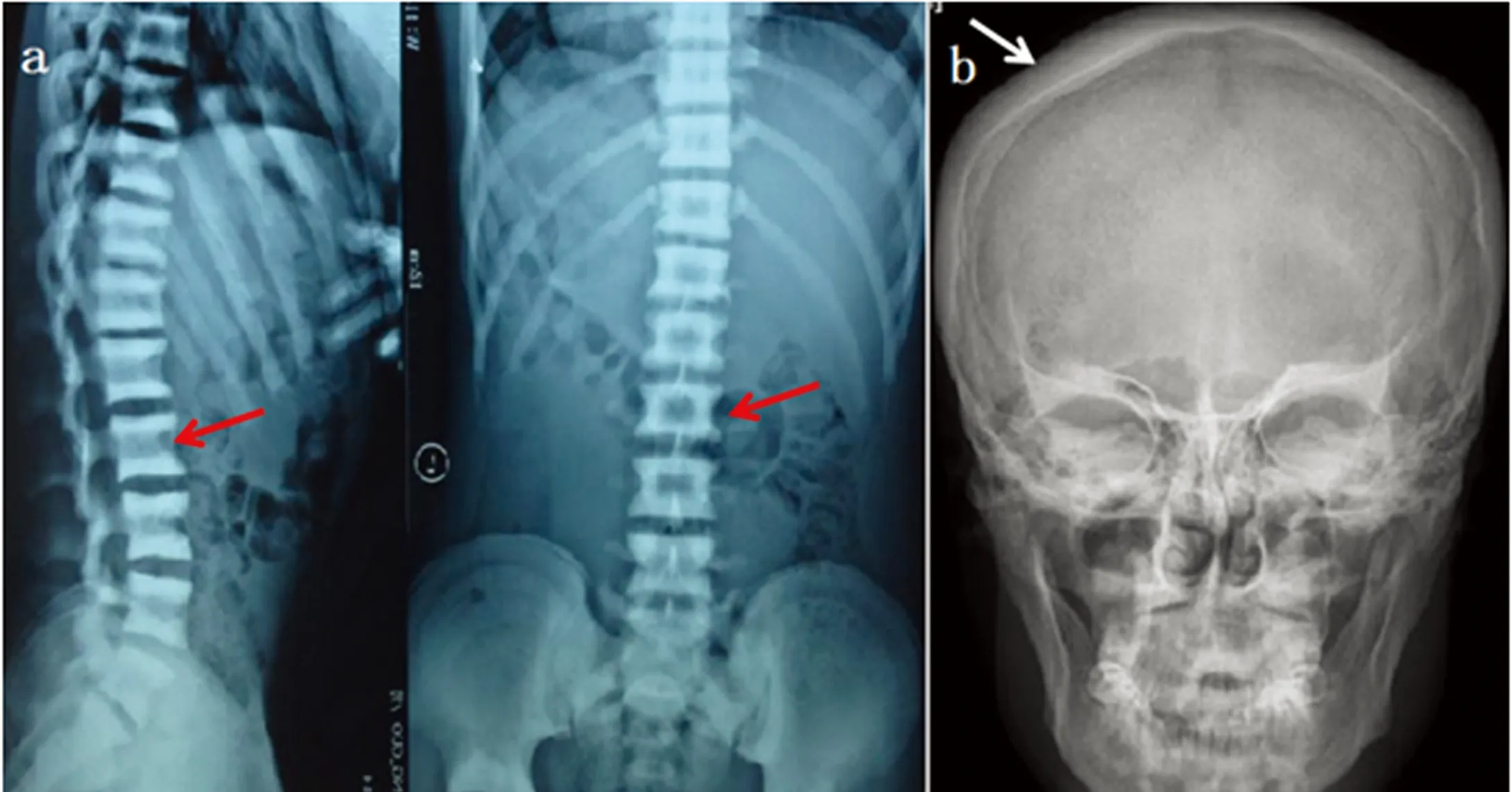
注:a.胸腰椎正側位相:椎體上下終板骨密度增厚,中央密度相對較低,呈“三明治”樣改變(紅色箭頭所示);b.頭顱正位相:顱骨骨皮質密度增厚,板障增厚,顱底骨質增厚硬化(白色箭頭所示)。
圖1先證者影像學特征
1.2血標本的采集及DNA的制備在遵循知情同意原則的基礎上,采集病人及其父母各2 mLEDTA抗凝血,用德國Qiagen全血DNA提取試劑盒提取病人及其家屬的外周血DNA。
1.3致病基因外顯子引物設計根據NCBI提供的人類基因組檢索網站(http://www.ncbi.nlm.nih.gov/gene),對人類野生型CLCN7的基因組序列進行檢索,使用專業生物學引物設計軟件GeneRunner software對該基因的編碼外顯子及其交界區進行PCR引物設計,經UCSC驗證,確保所有的引物都具有高度的特異性,擴增片段長度及退火溫度見表1。
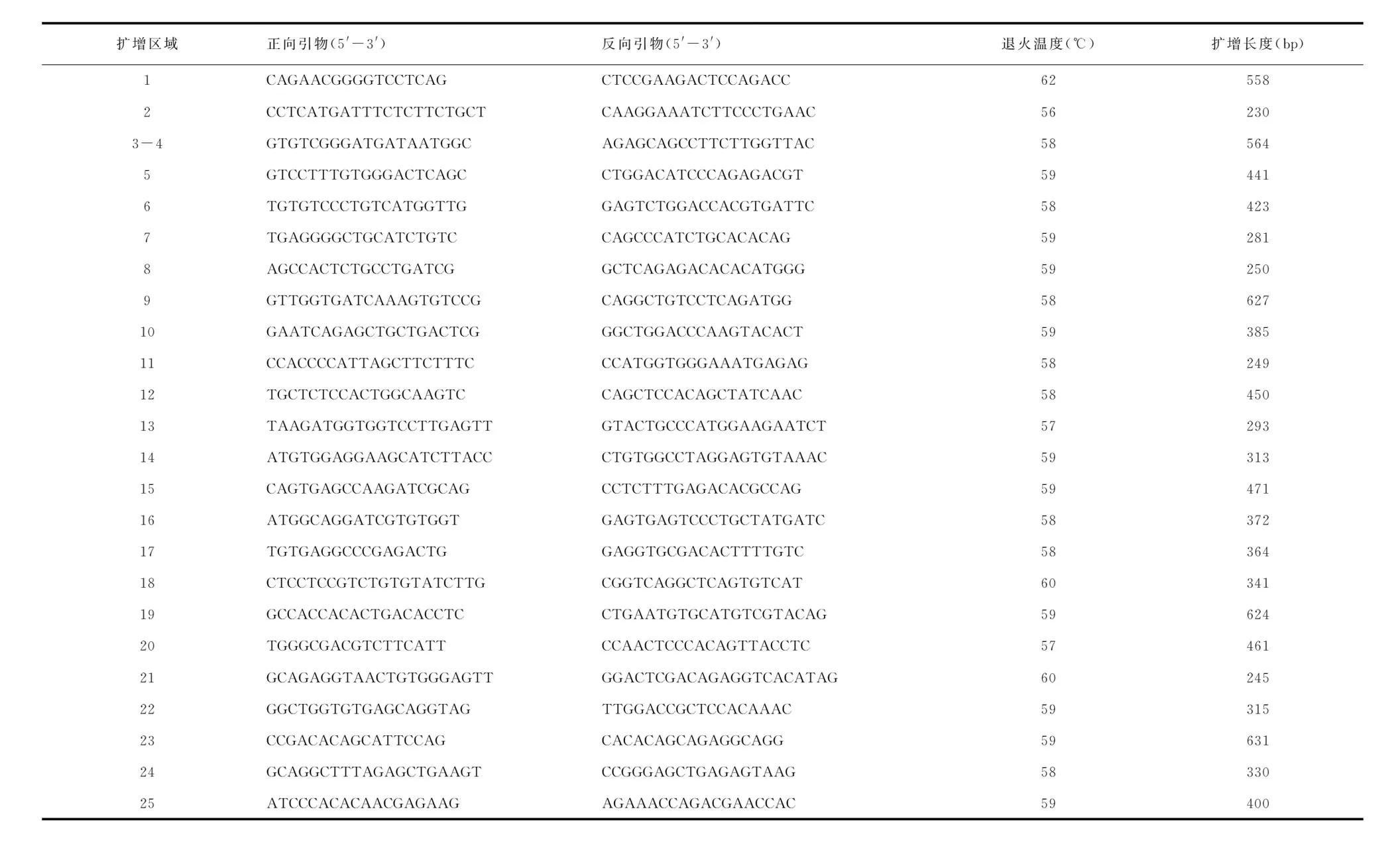
表1 CLCN7基因引物序列
1.4PCR擴增測序總反應體系30 μL:DNA模板2.6 μL,上下游引物各1.2 μL,2×Taq PCR 混合酶15 μL,ddH2O 10 μL;反應條件:預變性 95 ℃ 5 min,變性 94 ℃ 30 s,退火56 ℃ ~62 ℃ 30 s,延伸 72℃ 1 min,循環35輪,末次延伸 72℃ 10 min。
PCR產物測序:PCR產物送專業生物公司完成測序,使用美國A&B公司(Applied Biosystems,Foster City,CA)ABI3770 測序儀測序。
1.5DNA序列突變分析應用Chromas Version2.4軟件分析測序結果,將測序圖與正常的序列比對,然后應用UCSC和dbSNP138數據庫分別與基因組DNA序列進行比對。最后應用Human Gene Mutation Database Professional 2014(HGMD 2014)數據庫對突變體進行檢索,確定其是否為新發突變。對所有測序反應均雙向驗證。
2結果
該家系圖及測序結果見圖2。先證者及其父母CLCN7基因突變檢測結果示:先證者為第23外顯子c.2299C>T(p.R767W)的雜合錯義突變;其父母均非該突變的攜帶者,該突變為一自發突變。經查對2014HGMD突變數據庫,證實該突變為一已知突變,在2001年由Cleiren首次報道[5]。
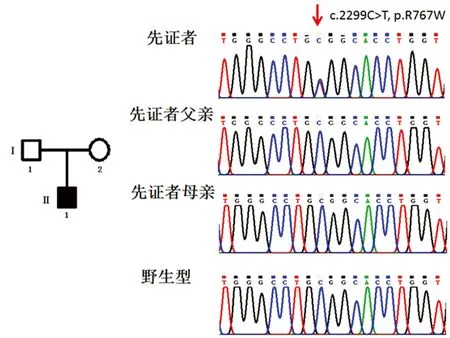
注:左邊為家系圖。黑色全黑方框為先證者(II1)。I1為先證者父親,I2為先證者母親,均非雜合子。右側為CLCN7基因測序圖。先證者為第23外顯子為c.2299C>T(p.R767W)的雜合錯義突變(箭頭所示),其父母親均非該突變攜帶者。
圖2病人家系圖及CLCN7基因測序結果
3討論
ADO-Ⅱ又名“Albers-Sch?nberg病”[6],是一種罕見的代謝性骨病,該亞型的臨床表現極度特異,且外顯率不全。ADO-Ⅱ型具有典型的ADO影像學特征,如椎體上下終板骨密度增厚,中央密度相對較低“夾心椎”樣表現及髂骨骨密度不均勻增高,出現濃淡相間的同心環狀的“骨中骨”現象。該病主要的并發癥體現在骨骼系統,包括病理性骨折、脊柱側凸、髖骨骨關節炎和上頜骨多發骨髓炎[6]。
目前認為編碼氯離子第7通道蛋白7基因突變是引起ADO-Ⅱ的主要原因[4]。CLCN7屬于多通道膜蛋白,主要功能是通過電壓-門控通道調節Cl-/H+交換。在破骨細胞中,CLCN7位于褶皺緣和溶酶體的隔室內,主要作用是將細胞外已通過離子交換進入細胞內的Cl-運輸入破骨細胞的小囊泡,在小囊泡中與通過質子泵運輸來的H+作用形成溶骨的酸性環境。破骨細胞吸收腔的低pH環境對于完成正常的骨吸收作用非常重要[7]。研究發現ADO-Ⅱ主要發病機制為CLCN7蛋白功能障礙,造成破骨細胞酸性環境形成受損,不足以發揮破骨細胞的骨吸收作用[4]。
本研究中先證者具有典型的骨硬化癥影像學表現,對該先證者進行CLCN7基因檢測,結果發現該病人CLCN7基因的第23外顯子c.2299C>T(p.R767W)的雜合錯義突變。該突變為一已知突變,但在國內罕有報道。Waguespack等[8]對62例ADO病人研究后發現,該R767W為較常見的一種。
本研究通過輔助檢查和基因檢測的方法,確證了1例中國人群罕有報道ADO-Ⅱ病人,進一步證實了CLCN7基因c.2299C>T (p.R767W)突變與該病人ADO-Ⅱ臨床表現密切相關。
參考文獻:
[1]Balemans W,Van Wesenbeeck L,VanHul W.A clinical and molecular overview of the human osteopetreses [J].Calcif Tissue Int,2005,77: 263-274.
[2]柯耀華,章振林.骨硬化癥致病基因研究進展[J].中華骨質疏松和骨礦鹽疾病雜志,2010,3(2): 1674-2591.
[3]Andrea Del Fattore,Alfredo Cappariello,Anna Teti.Genetics,pathogenesis and complications of osteopetrosis[J].Bone,2008,42: 19-29.
[4]Kasper D,Planells-Cases R,Fuhrmann JC,et al.Loss of the CLC-7 chloride channel leads to osteopetrosis in mice and man [J] Cell,2001,104: 205-215.
[5]Cleiren E,Benichou O,Van Hul E,et al.lbers-Schonberg disease (autosomal dominant osteopetrosis,type Ⅱ) results from mutations in the ClCN7 chloride channel gene[J].Hum Mol Genet,2001,10: 2861-2867.
[6]Zornitza Stark,Ravi Savarirayan.Osteopetrosis[J].Orphanet Journal of Rare Diseases,2009,4:5.
[7]Graves AR,Curran PK,Smith CL,et al.The Cl(/H)(+)antiporter ClC-7 is the primary chloride permeation pathway in lysosomes[J].Nature,2008,453: 788-792.
[8]Waguespack SG,Koller DL,White KE,et al.Chloride channel 7(ClCN7) gene mutations and autosomal dominant osteopetrosis,type Ⅱ[J].J Bone Miner Res,2003,18: 1513-1518.
(本文編輯王雅潔)
通訊作者:董進,E-mail: Sdyydj@medmail.com.cn
中圖分類號:R596
文獻標識碼:C
doi:10.3969/j.issn.1672-1349.2016.13.048
文章編號:1672-1349(2016)13-1566-03
(收稿日期:2015-05-30)
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
