脂肪變肝細胞中炎性調節蛋白A20的表達機制
2016-08-11 01:23:46許青青艾羅燕吳昌維蘇大芝王曉晗陳志威江小柯范竹萍
國際消化病雜志 2016年3期
關鍵詞:信號
許青青 艾羅燕 吳昌維 蘇大芝 王曉晗 陳志威 江小柯 林 卿 范竹萍
?
脂肪變肝細胞中炎性調節蛋白A20的表達機制
許青青艾羅燕吳昌維蘇大芝王曉晗陳志威江小柯林卿范竹萍
200001上海交通大學醫學院附屬仁濟醫院健康保健中心,上海市消化疾病研究所(許青青,艾羅燕,蘇大芝,王曉晗,陳志威,江小柯,范竹萍);475000開封,河南大學附屬淮河醫院感染科(吳昌維);201203桑迪亞醫藥技術(上海)有限責任公司(林卿)
非酒精性脂肪性肝病(NAFLD)已被認為是一種慢性低度炎性反應狀態[1],炎性反應在其復雜的發病機制中至關重要,是疾病向終末期肝病進展的關鍵。鋅指蛋白A20是由核因子-κB(NF-κB)激活誘導的炎性反應負性調節蛋白,它通過可逆的去泛素化以及泛素化功能下調活化的NF-κB信號通路,從而抑制炎性反應[2]。近年來研究發現,A20對肝臟再生及功能恢復具有積極作用,可抑制肝損傷、肝移植、缺血再灌注等誘發的炎性反應[3-4],但是否對NAFLD有調控作用未見報道。前期研究已采用油酸和軟脂酸比為2∶1的混合脂肪酸(FFA)作用HepG2細胞建立脂肪變模型,發現隨著FFA濃度升高,A20的表達有時間限定性并呈現出一定的規律,推測A20可能是通過抑制NF-κB信號通路激活來抑制炎性反應的發生,從而改善NAFLD[5],本研究在此基礎上進一步探討A20表達時信號通路的激活情況。
1 材料與方法
1.1材料
肝癌細胞株HepG2由桑迪亞醫藥技術(上海)有限責任公司惠贈。主要試劑:DMEM高糖培養基、胎牛血清、0.25%胰酶(Gibco公司),二甲基亞砜(DMSO)、油酸鈉、軟脂酸鈉(Sigma公司),三酰甘油(TG)試劑盒(南京建成生物工程研究所),RIPA(中)裂解液(碧云天生物技術研究所),兔抗人A20抗體、GAPDH抗體、HRP-偶聯的羊抗兔IgG(Cell Signaling Technology公司),ECL化學發光試劑盒(Perkinelmer公司),人細胞因子檢測試劑盒(BD公司)。其余試劑均為國產或進口分析純。
1.2方法
1.2.1細胞培養及對照組取(3×105~5×105)/mL HepG2細胞,以含10%胎牛血清的DMEM培養液培養,置于37 ℃、含5%CO2的培養箱中孵育,隔天換液,并按1∶5傳代培養。對照組用DMEM培養液培養細胞,無混合脂肪酸(FFA)母液。
1.2.2FFA配制分別取304.4 mg油酸和128.2 mg軟脂酸,溶于10 mL甲醇,配成150 mmol/L FFA母液。根據實驗需要,將母液按比例稀釋成相應工作液,DMSO濃度為0.5%。
1.2.3細胞內TG含量測定將HepG2細胞按2×105/mL接種于6孔板中,常規培養24 h后更換培養液,用0.5 mmol/L FFA分別作用于HepG2細胞0、0.25、0.5、1、2、4、6、12、24 h;將6孔培養板中培養液吸出,冰PBS洗滌細胞2次,加入RIPA裂解液30 μL(含蛋白酶抑制劑),后置于冰上裂解30 min,刮出并轉移至Eppendorf管;4 ℃,12 000 rpm離心15 min,小心吸取上清,轉移至Eppendorf管;按照TG試劑盒說明書進行操作,測定細胞內的TG含量。
1.2.4細胞內蛋白表達檢測將HepG2細胞2×105/mL接種于6孔板,次日加入0.5 mmol/L FFA作用6、12、24 h后,加入細胞裂解液,收取蛋白樣品行SDS-PAGE電泳,濕法電轉移法轉移到NC膜,5%脫脂牛奶封閉2 h,分別加入A20(1∶1 000稀釋)和GAPDH(1∶10 000稀釋),4 ℃搖床孵育過夜,二抗(1∶20 000稀釋)室溫孵育1 h。ECL發光劑顯色后,置暗盒曝光于X膠片上,常規顯影、定影后,掃描條帶,用Quality One分析軟件半定量結果,目的蛋白表達量=目的蛋白條帶灰度值/GAPDH條帶灰度值。
1.2.5細胞上清中白細胞介素-8含量測定收集相應的細胞上清100 μL,2 mL標準品稀釋液加到標準品中溶解,室溫靜置15 min;制定標準曲線,取6個bead各2 μL/樣品,PE director各12 mL/樣品,兩者振蕩混勻;96孔“V”型板中,加入24 μL上述混合物;加入樣品或各個濃度標準品12 μL/孔,上下吹勻;25 ℃,150 rpm,避光孵育3 h;加入150 μL/孔wash buffer,200 g,離心5 min;甩掉液體,200 μL/孔wash buffer;轉移至流式管中,最后流式細胞儀上機操作(按說明書設立程序進行)。
1.3統計學分析
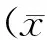
2結果
2.1細胞內脂質沉積
0.5 mmol/L FFA在0~24 h不同時間點作用于HepG2細胞后,測得細胞內TG含量0時為(0.07±0.00)mmol/L,隨著作用時間的延長,從4 h開始增加,12 h到達峰值(0.13±0.00)mmol/L(P<0.05)(見圖1)。
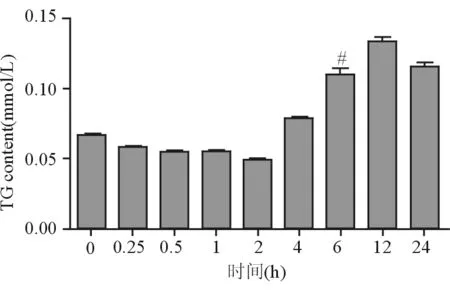
注:與對照組相比較,#P<0.05
圖1細胞內TG含量
2.2細胞內蛋白表達水平
0.5 mmol/L FFA在0~24 h不同時間點作用于HepG2細胞時,發現各個時間點A20的表達不同,呈波浪狀起伏,與對照組相比,A20、pIκB在0.25、1、4、6、12 h時表達升高,活化的p65(p65p)在4、6、12 h時表達顯著升高,表明FFA介導NF-κB信號通路活化,A20被誘導呈瞬時表達(見圖2、圖3)。
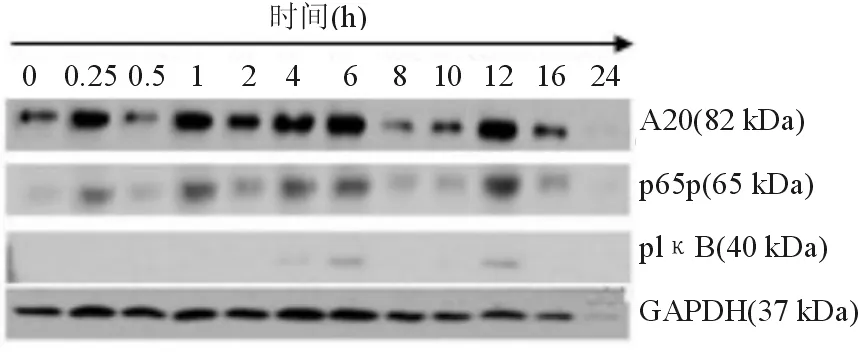
注:1Da=0.9921 u
圖2HepG2細胞在不同時間點的蛋白A20、p65p、pIκB表達
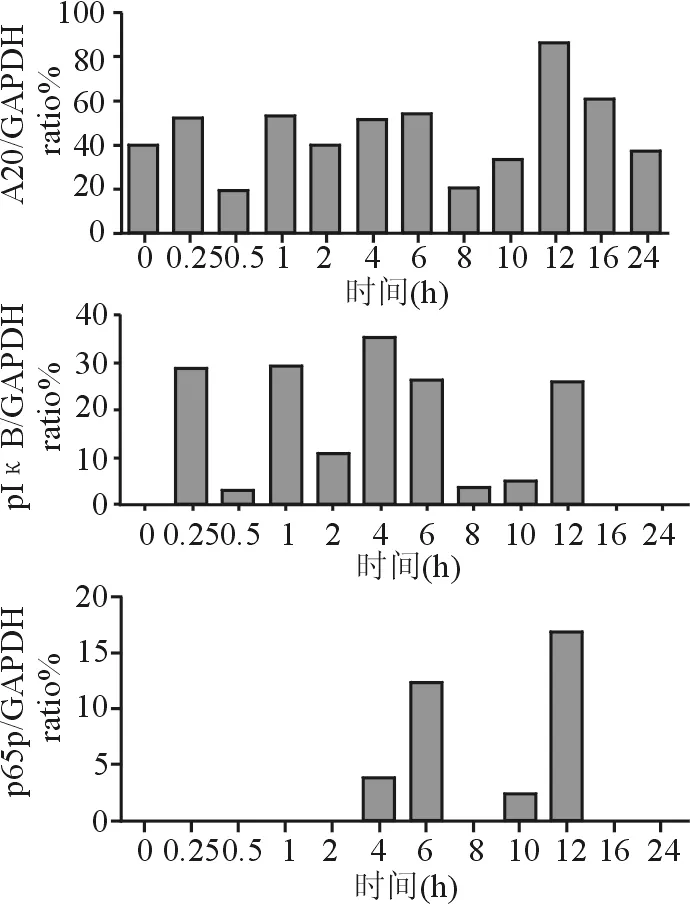
圖 3 HepG2細胞在不同時間點的蛋白A20、p65p、pIκB半定量結果
2.3細胞上清中白細胞介素-8含量
0.5 mmol/L FFA在0~24 h不同時間點作用于HepG2細胞時,與對照組(9.57±1.95)相比,在4、6、12、24 h時細胞上清中的白細胞介素-8(IL-8)水平顯著升高[(69.83±3.65)、(95.20±3.95)、(51.34±3.45)、(35.57±3.15) ng/mL](P<0.05),分泌時相呈先升高后降低趨勢,6 h時為峰值(見圖4)。
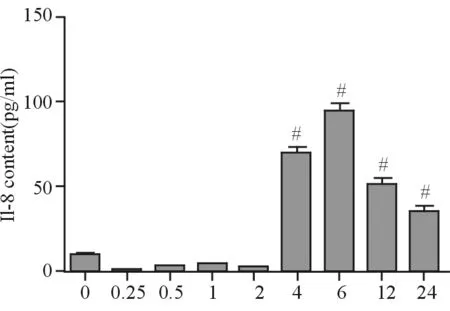
注:與對照組相比,#P<0.05
圖4HepG2細胞在不同時間點的細胞上清中IL-8含量
3討論
NAFLD是一種慢性低度炎性反應狀態已成為國際共識,炎性反應是其進展的重要原因[6],這種炎性反應不僅可以加重胰島素抵抗,影響營養物質代謝,還使得原本靜息的肝星狀細胞活化,促進了肝纖維化、肝硬化的發生,因此抑制炎性反應對阻斷疾病進展具有重要意義。NF-κB通路是此慢性炎性反應發生的主要信號通路[7],NF-κB是一個異源二聚體蛋白(p50/p65),在大部分細胞中處于失活狀態,被其抑制蛋白(IκB)禁錮在細胞質中,只有在刺激信號的作用下,IκB在IκB激酶復合體(IKK)的作用下快速發生磷酸化修飾形成pIκB,而后被26S蛋白酶體降解,從而釋放 NF-κB蛋白,p65p發生核轉移,啟動一系列的炎性反應基因轉錄。鋅指蛋白A20是由NF-κB通路激活誘導的炎性反應負性調節蛋白,具有雙重泛素編輯酶活性,通過抑制轉錄因子NF-κB 的活化,阻斷炎性反應持續發生及炎性因子表達,從而廣泛參與炎性反應及免疫的調節[8-9]。那么,A20對NAFLD是否有調控作用成為本研究最關心的問題。
本研究通過原先建立的NAFLD細胞模型,在此基礎上進一步利用0.5 mmol/L FFA在不同時間點作用于HepG2細胞,細胞內TG含量隨時間延長而增加,細胞上清中IL-8水平在4 h時顯著升高,峰值出現在6 h,其后逐漸降低,呈現脈沖相表達,提示FFA能誘發脂肪沉積和炎性反應。這與以往研究報道一致,如Li等[10]發現FFA通過作用于Toll樣受體(TLR)內源性配體,激活IKK/NF-κB、JNK/AP-1信號通路,誘導NAFLD形成;同樣Joshi-Barve等[11]用軟脂酸刺激肝細胞(HepG2、鼠原代肝細胞、人原代肝細胞)后,NF-κB、JNK/AP-1信號通路激活,IL-8水平升高。此外,本研究觀察到NF-κB信號通路的激活:pIκB和p65p在4、6、12 h時表達顯著升高,IL-8水平亦升高,隨之A20表達升高,并呈波浪樣的改變。這符合A20表達有明顯的時效性特點,只要外界刺激能激活NF-κB,活化的NF-κB轉移至細胞核內與A20啟動子區的兩個識別序列的κB元件結合,就能迅速啟動A20基因的轉錄,而表達升高是機體的一種重要的內源性抗炎保護效應機制,可能對于調控肝內慢性低度炎性反應具有重要意義。有研究發現,肝內高表達A20后,大量參與脂代謝通路的基因和線粒體內膜多種調節代謝的基因表達上調,如脂肪酸、氨基酸、膽固醇及類固醇的合成基因表達升高,使脂代謝增強,加快能量輸出,還影響外周脂肪流動、肝內脂肪酸攝取,肝細胞內TG快速蓄積等代謝過程,說明A20對肝臟的保護作用不僅僅局限于抑制損傷、移植及缺血再灌注所誘發的炎性反應,還具有調節脂質代謝、產生能量的作用[12]。可見A20在肝臟中的多方面作用正被逐漸發掘和廣泛接受[13]。A20與炎性反應、代謝的關系及機制研究也備受關注,其在NAFLD疾病發展中的作用值得深入探討。
本研究在HepG2細胞脂肪變模型中觀察到FFA能誘導NF-κB信號通路激活,IL-8水平升高,A20負反饋表達升高,提示可上調A20的表達以加強機體固有免疫來發揮抗炎作用,為NALFD的防治提供可能的有效作用靶點。然而本實驗中A20與NAFLD之間的關系探討僅局限于細胞水平,更多深入的分子機制以及動物水平的表達規律有待進一步研究。
志謝感謝桑迪亞醫藥技術(上海)有限責任公司生物部各位老師在操作規范和實驗技術上給予的指導
參考文獻
1 Medina-Santillán R, López-Velázquez JA, Chávez-Tapia N, et al. Hepatic manifestations of metabolic syndrome[J]. Diabetes Metab Res Rev, 2013, Mar 7. [Epub ahead of print]
2 Harhaj EW, Dixit VM. Regulation of NF-κB by deubiquitinases[J]. Immunol Rev, 2012, 246: 107-124.
3 Studer P, da Silva CG, Revuelta Cervantes JM, et al. Significant lethality following liver resection in A20 heterozygous knockout mice uncovers a key role for A20 in liver regeneration[J]. Cell Death Differ, 2015, 22: 2068-2077.
4 da Silva CG, Studer P, Skroch M, et al. A20 promotes liver regeneration by decreasing SOCS3 expression to enhance IL-6/STAT3 proliferative signals[J]. Hepatology, 2013, 57: 2014-2025.
5 許青青, 吳昌維, 艾羅燕, 等. 游離脂肪酸誘導肝細胞脂肪變模型中鋅指蛋白A20的表達及其作用機制[J]. 胃腸病學, 2013, 18: 95-99.
6 Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis[J]. Hepatology, 2010, 52: 1836-1846.
7 Marra F. Nuclear factor-kappaB inhibition and non-alcoholic steatohepatitis: inflammation as a target for therapy[J]. Gut, 2008, 57: 570-572.
8 Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity[J]. Nature, 2009, 458: 430-437.
9 Sriskantharajah S, Ley SC. Cell biology. Turning off inflammation signaling[J]. Science, 2010, 327: 1093-1094.
10 Li LL, Chen L, Hu L, et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice[J]. Hepatology, 2011, 54: 1620-1630.
11 Joshi-Barve S, Barve SS, Amancherla K, et al. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes[J]. Hepatology, 2007, 46: 823-830.
12 Damrauer SM, Studer P, da Silva CG, et al. A20 modulates lipid metabolism and energy production to promote liver regeneration[J]. PLoS One, 2011, 6: e17715.
13 da Silva CG, Cervantes JR, Studer P, et al. A20--an omnipotent protein in the liver: prometheus myth resolved [J]. Adv Exp Med Biol, 2014, 809: 117-139.
(本文編輯:林磊)
通信作者:范竹萍,Email: zhuping_fan@163.com
DOI:10.3969/j.issn.1673-534X.2016.03.017
(收稿日期:2015-12-20)
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
