擔體類型對鈷基催化材料的CO加氫性能與活化能的影響
2016-09-02 10:04:34劉振新薛瑩瑩吳德鵬方少明
功能材料 2016年5期
關鍵詞:催化劑
邢 宇,劉振新,薛瑩瑩,吳德鵬,方少明
(鄭州輕工業學院 材料與化學工程學院,河南省表界面科學重點實驗室,鄭州 450002)
?
擔體類型對鈷基催化材料的CO加氫性能與活化能的影響
邢宇,劉振新,薛瑩瑩,吳德鵬,方少明
(鄭州輕工業學院 材料與化學工程學院,河南省表界面科學重點實驗室,鄭州 450002)
在450~550 ℃的還原溫度下,Co/ZnO催化材料存在2種結晶態鈷物種。在空速為1 500mL/g催化劑·小時、總壓為2.0MPa、H2∶CO比為2∶1的反應條件下,對比了負載在路易斯堿性擔體(ZnO)和路易斯酸性擔體(γ-Al2O3)上的鈷基催化材料的費托合成催化性能,發現不同酸堿類型的擔體對鈷基催化材料的CO加氫活性、產物選擇性以及表觀活化能均有顯著的影響。
鈷;催化材料;復合材料;擔體;CO加氫
0 引 言
鈷系復合催化材料可以用在有機物加氫[1-2]、催化燃燒[3]、光催化[4]等領域,然而,鈷系催化材料更為重要的應用是在CO加氫的費托合成領域[5-16]。費托合成是將合成氣(CO+H2)轉化成烴類及含氧有機化合物的化學過程,是非石油含碳資源(天然氣、煤炭、渣油以及生物質等) 高效轉化利用最重要的途徑之一。合成氣在使用不同催化劑和不同工況條件下,可以選擇性生成一系列不同碳數的高級烴(C1~C200),主要是直鏈烷烴,也可以選擇性生成低碳烯烴,還可以選擇性生成醇類等產品,適合我國富煤少油的實際國情[6-10]。使用鈷系催化材料時產生的費托合成初產品經進一步處理(如分離、加氫裂解或者加氫異構化等) ,可以得到一定規格的汽油、柴油等油品燃料以及乙烯、丙烯、潤滑油和石蠟等化學品[7]。鈷系催化材料在費托合成中的反應已有較多研究文獻[5-16],但是擔體酸堿類型對于鈷基催化材料的CO加氫性能與活化能的研究則很少[17]。ZnO為路易斯堿性擔體[18],而γ-Al2O3為路易斯酸性擔體[19]。本文在空速為1 500mL/克催化劑·小時、總壓為2.0MPa、H2∶CO比為2∶1的反應條件下,對比了負載在路易斯堿性擔體上的鈷基催化材料(Co/ZnO)和路易斯酸性擔體上的鈷基催化材料(Co/γ-Al2O3)的費托合成催化性能,測定了表觀活化能,發現不同酸堿類型的擔體對催化性能和表觀活化能有著顯著的影響。
1 實 驗
1.1合成
Co/ZnO的制備:硝酸鋅水溶液與沉淀劑NaHCO3水溶液在40 ℃的劇烈攪拌中實施沉淀,然后在40 ℃的劇烈攪拌中陳化1h,過濾后多次充分洗濾,濾餅在80 ℃部分脫水,摻入少量助擠劑(田菁粉),捏合后擠條,晾干,110 ℃烘干2h,再于350 ℃煅燒4h,破碎后過40~60目篩,生成ZnO擔體;用硝酸鈷水溶液浸漬該ZnO擔體,烘干后在350 ℃煅燒4h,制得“未還原的Co/ZnO”;用50%H2/50%Ar氣氛在常壓下還原后制得Co/ZnO,本文中的還原溫度分別為450,500和550 ℃,所得催化材料分別標記為Co/ZnO-450、Co/ZnO-500、Co/ZnO-550。Co/γ-Al2O3也用浸漬法制備,即用硝酸鈷水溶液浸漬γ-Al2O3擔體,還原溫度為550 ℃,具體細節見我們近期的論文[17]。Co/ZnO和Co/γ-Al2O3催化材料的Co元素負載量均為15%(質量分數)。Co元素在Co/ZnO和Co/γ-Al2O3催化材料的實際含量(ICP法)分別為14.85%和15.10%(質量分數)。
1.2表征
樣品結構用PanalyticalX’PertPro粉末X射線衍射儀測定,化學組成用Perkin-ElmerElan9000型電感耦合等離子體質譜儀測定,樣品形貌用HitachiS-4700場發射電子顯微鏡和FEITecnaiG20TEM透射電子顯微鏡觀察。平均晶粒尺寸的計算與表示方法如文獻[17]所述。樣品的比表面積用QuantachromeNOVA1000表面積與孔徑分析儀測定。
1.3費托反應條件
反應裝置為天津市天大北洋化工實驗設備有限公司生產的固定床費托合成裝置。新鮮制備的未還原鈷系催化材料裝入反應器后,先在550 ℃用50%H2/50%He混合氣原位還原,然后切換至預混合的合成氣(H2/CO/Ar=6/3/1(摩爾分數))并緩慢升壓至2.0MPa,溫度緩慢升至所需測定的反應溫度實施費托合成反應,空速為1 500mL/克催化劑·小時。產物用氣相色譜儀分析[17]。
2 結果與討論
2.1材料的制備與表征
圖1為Co/ZnO催化材料的X-射線衍射譜圖。

圖1 Co/ZnO催化材料的X-射線衍射譜圖
Fig1X-raydiffraction(XRD)patternsofCo/ZnOcatalyticmaterials
如圖1所示,未加標記的XRD衍射峰歸屬于六方ZnO相(JCPDS: 36-1451)。在450,500和550 ℃還原所得的Co/ZnO樣品中ZnO相的平均晶粒尺寸(LZnO(101),指垂直于ZnO(101)晶面的方向上的ZnO晶體平均厚度;其它可類推)分別為114.1,205.0和1 145.0nm。Co/ZnO樣品中含有立方Co0相(JCPDS: 15-806)和六方Co(OH)2相(JCPDS: 30-0443)。樣品Co/ZnO-550中Co0相的平均晶粒尺寸(LCo(111))為31.9nm。樣品Co/ZnO-450中Co(OH)2相的衍射峰很弱, 說明在450 ℃的還原中該相結晶度很低。樣品Co/ZnO-500中Co(OH)2相的衍射峰比樣品Co/ZnO-450中的要強一些, 說明在500 ℃的還原中該相結晶度比在450 ℃更高,這可能是Co(OH)2晶體在500 ℃因燒結而長大的程度大于450 ℃。樣品Co/ZnO-550中Co(OH)2相的衍射峰比樣品Co/ZnO-500的明顯要弱一些,而且樣品Co/ZnO-550中Co0相的衍射峰比樣品Co/ZnO-500的明顯要強一些,說明在550 ℃ 的較高還原溫度下Co(OH)2相可以被逐漸還原為Co0相。我們在Co/γ-Al2O3的XRD研究中僅發現了結晶態Co0相和結晶態γ-Al2O3相,沒有發現過結晶態Co(OH)2相[17]。這說明結晶態Co(OH)2相的出現與ZnO有關。樣品Co/ZnO-550中,Co0(111)衍射峰與Co(OH)2(101)衍射峰的峰強度之比約為1∶0.9。
圖2(a)-(c)所示為樣品Co/ZnO-550的FESEM圖像。這些圖像說明樣品Co/ZnO-550由兩種不同尺寸范圍的顆粒所組成,一種顆粒的尺寸處于500~1 000nm,另一種顆粒的尺寸處于20~50nm;大顆粒的前者應該屬于ZnO相,小顆粒的后者應該屬于鈷物種。小顆粒易在大顆粒的表面上聚集。圖2(d)所示為樣品Co/γ-Al2O3的FESEM圖像,說明構成該樣品的顆粒尺寸很小,僅為10nm左右,但是團聚現象比較明顯。

圖2催化材料的FESEM圖像
Fig2FESEMimagesofcatalyticmaterials
圖3所示為Co/ZnO-550的TEM圖像,說明鈷物種在ZnO擔體上的分布并不十分均勻。樣品Co/γ-Al2O3的BET比表面積為128.6m2/g;樣品Co/ZnO-550的BET比表面積為2.8m2/g。假設Co0晶體的形狀為立方體,且定義分散度為晶粒表面上的Co原子數占催化劑中Co原子總數的比例,根據樣品Co/γ-Al2O3和Co/ZnO-550的Co0相平均晶粒尺寸(LCo(111))分別為12.1和31.9nm,可以算出樣品Co/γ-Al2O3和Co/ZnO-550的Co分散度分別為9.9%和3.8%。

圖3Co/ZnO催化材料的TEM圖像
Fig3TEMimagesofCo/ZnOcatalyticmaterial
2.2CO加氫催化性能與表觀活化能

300 ℃時Co/ZnO-550的轉化率僅為0.7%,而用來檢測CO2和CH4產物的TCD檢測器靈敏度則有限,且CH4的選擇性又是關聯其它烴類選擇性的基礎,因此300 ℃時Co/ZnO-550的各個產物選擇性數據因為準確度不足而不能使用。310~370 ℃時Co/ZnO-550的各個產物選擇性數據可用。一般認為CO加氫過程中CO2主要是由水汽變換(water-gasshift,WGS)反應產生的[20]。酸性擔體負載的鈷催化材料(Co/γ-Al2O3)的CO2選擇性很低,說明金屬Co和酸性擔體γ-Al2O3對水汽變換都是不活潑的。堿性擔體負載的鈷催化材料(Co/ZnO-550)的CO2選擇性較高,說明堿性擔體ZnO對水汽變換是活潑的。由于CO*不能直接與穩定的H2O*發生反應,因此水汽變換反應始自水的離解過程中OH*、O*和H*的生成[21]。ZnO路易斯堿可以化學吸附H2O并使之發生異裂[22]。這可能是Co/ZnO-550具有較高水汽變換活性的原因。

圖4是用阿累尼烏斯曲線法測定的表觀活化能。在空速為1 500mL/克催化劑·小時、總壓為2.0MPa、H2∶CO比為2∶1的反應條件下,酸性擔體負載的鈷催化材料(Co/γ-Al2O3)上CO轉化的表觀活化能為120.1kJ/mol,堿性擔體負載的鈷催化材料(Co/ZnO-550)上CO轉化的表觀活化能為165.6kJ/mol。

圖4 阿累尼烏斯曲線法測定表觀活化能
Fig4Roughdeterminationofapparentactivationenergyvaluesusingarrheniusplots
酸性擔體負載的鈷催化材料的表觀活化能低于堿性擔體負載的鈷催化材料,這與酸性擔體負載的鈷催化材料的CO加氫活性高于堿性擔體負載的鈷催化材料的現象是一致的。我們將在以后的研究中考察純相Co(OH)2的催化性能,以探索不同價態的Co對催化結果的影響。此外,SiO2與Al2O3相比亦具有代表性,因此我們將會考察SiO2擔體負載的鈷催化材料并加以對比。
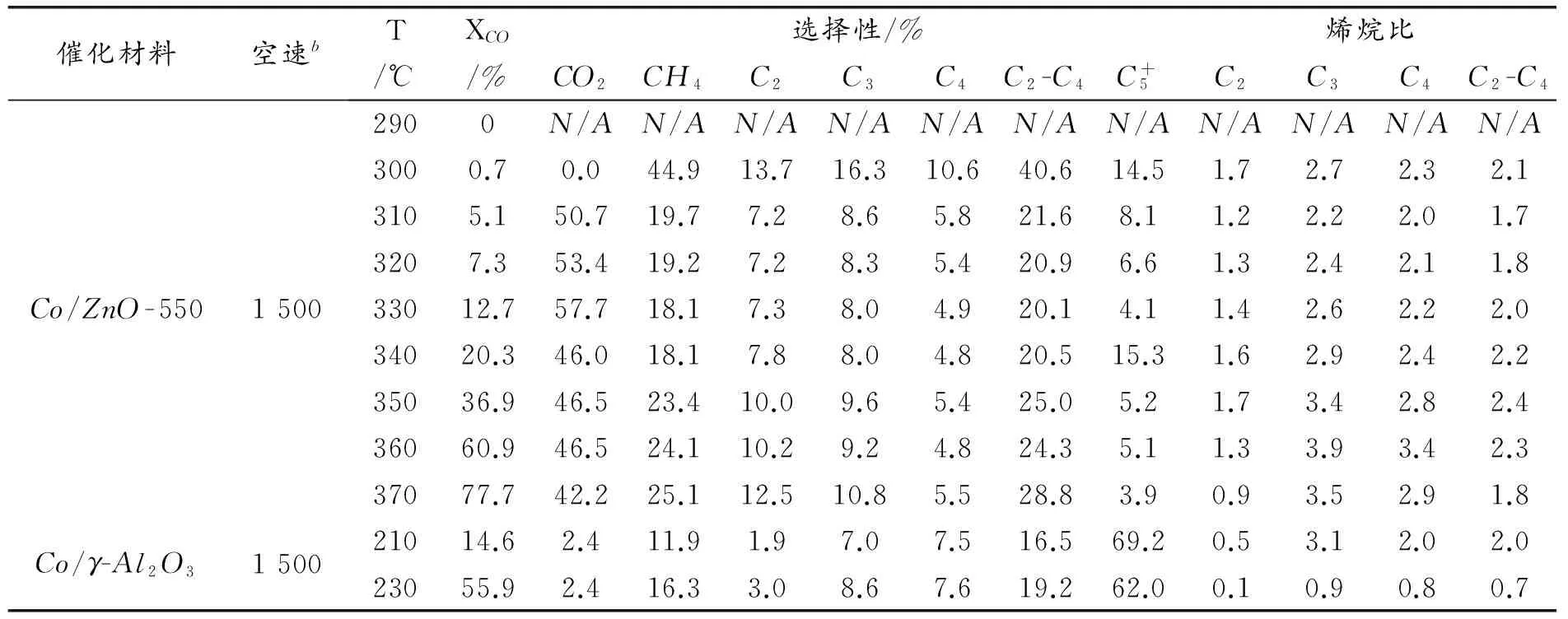
表1 催化材料的CO加氫催化結果a
a反應條件:P總=2.0MPa,H2/CO/Ar=6/3/1(摩爾比)。數據均基于C元素的摩爾數。b空速(SV)的單位是[毫升/克催化劑·小時]。
3 結 論
[1]LuS,MenningCA,ZhuY,etal.CorrelatingbenzenehydrogenationactivitywithbindingenergiesofhydrogenandbenzeneonCo-basedbimetalliccatalysts[J].ChemPhysChem, 2009, 10: 1763-1765.
[2]ZhangXB.Preparationof1-phenylethanolbyselectivehydrogenationofacetophenoneoveralumina-supportedCocatalysts[J].ReacKinetMechCat, 2011, 102: 417-424.
[3]WangL,LiX,XiaQ,etal.Catalyticcombustionoftolueneovercobaltbasedcatalystspreparedbychemicalreductionwithethyleneglycol[J].JournalofFunctionalMaterials, 2012, 43: 835-838.
王麗間, 李欣, 夏啟斌, 等. 乙二醇法制備負載型鈷基-堇青石催化劑及其催化燃燒甲苯的活性[J]. 功能材料, 2012, 43: 835-838.
[4]ChenQ,JiF,GuoQ,etal.MechanismofCo-TiO2catalystwithbothvisiblelightphotocatalyticandFenton-likecatalyticactivities[J].JournalofFunctionalMaterials, 2013, 24: 3616-3621.
陳晴空, 吉芳英, 郭倩,等. 兼具光催化和類Fenton活性的Co-TiO2催化機理研究[J]. 功能材料, 2013, 24: 3616-3621.
[5]HuangW,WangJ,SunZ,etal.EffectofreductiontemperatureonperformanceofdoublemesoporousCo-basedcatalystsinFischer-Tropschsynthesis[J].JournalofFuelChemistryandTechnology, 2014, 42: 81-86.
黃巍, 王俊剛, 孫志強,等. 還原溫度對雙介孔鈷基催化劑費-托合成性能的影響[J]. 燃料化學學報, 2014, 42: 81-86.
[6]JahangiriH,BennettJ,MahjoubiP,etal.AreviewofadvancedcatalystdevelopmentforFischer-Tropschsynthesisofhydrocarbonsfrombiomassderivedsyn-gas[J].CatalSciTechnol, 2014, 4: 2210-2229.
[7]KrylovaAY.PruductsoftheFischer-Tropschsynthesis[J].SolidFuelChem, 2014, 48: 22-35.
[8]ZhangQ,ChengK,KangJ,etal.Fischer-Tropschcatalystsfortheproductionofhydrocarbonfuelswithhighselectivity[J].ChemSusChem,2014, 7: 1251-1264.
[9]JinE,ZhangY,HeL,etal.Indirectcoaltoliquidtechnologies[J].ApplCatalA-Gen, 2014, 476: 158-174.
[10]ZhaiP,SunG,ZhuQ,etal.Fischer-Tropschsynthesisnanostructuredcatalysts:understandingstructuralcharacteristicsandcatalyticreaction[J].NanotechnolRev, 2013, 2: 547-576.
[11]SartipiS,MakkeeM,KapteijnF,etal.Catalysisengineeringofbifunctionalsolidsfortheone-stepsynthesisofliquidfuelsfromsyngas:areview[J].CatalSciTechnol, 2014, 4: 893-907.
[12]LiuY,ErsenO,MenyC,etal.Fischer-Tropschreactiononathermallyconductiveandreusablesiliconcarbidesupport[J].ChemSusChem,2014, 7: 1218-1239.
[13]BeaumontSK.RecentdevelopmentsintheapplicationofnanomaterialstounderstandingmolecularlevelprocessesincobaltcatalysedFischer-Tropschsynthesis[J].PhysChemChemPhys, 2014, 16: 5034-5043.
[14]YangJ,MaW,ChenD,etal.Fischer-Tropschsynthesis:areviewoftheeffectofCOconversiononmethaneselectivity[J].ApplCatalA-Gen, 2014, 470: 250-260.
[15]MosayebiA,HaghtalabA.ThecomprehensivekineticmodelingoftheFischer-TropschsynthesisoverCo@Ru/γ-Al2O3core-shellstructurecatalyst[J].ChemEngJ, 2015, 259: 191-204.
[16]ZhangQ,KangJ,WangY.DevelopmentofnovelcatalystsforFischer-Tropschsynthesis:tuningtheproductselectivity[J].ChemCatChem,2010, 2: 1030-1058.
[17]LiuZ,XingY,XueY,etal.Synthesis,characterizationandFischer-Tropschperformanceofcobalt/zincaluminatenanocompositesviaafacileandcorrosion-freecoprecipitationroute[J].JNanopartRes, 2015, 17:DOI: 10.1007/s11051-015-2899-3
[18]BuscaG.Basesandbasicmaterialsinchemicalandenvironmentalprocesses,liquidversussolidbasicity[J].ChemRev, 2010, 110: 217-2249.
[19]MaitlisPM,ZanottiV.TheroleofelectrophilicspeciesintheFischer-Tropschreaction[J].ChemCommun, 2009,1619-1634.
[20]OjedaM,NabarR,NilekarAU,etal.COactivationpathwaysandthemechanismofFischer-Tropschsynthesis[J].JCatal, 2010, 272: 287-297.
[21]LinCH,ChenCL,WangJH.Mechanisticstudiesofwater-gas-shiftreactionontransitionmetals[J].JPhyChemC, 2011, 115: 18582-18588.
[22]KokesRJ.Hydrogenationandisomerizationoverzincoxide[J].AdvCatal, 1972, 1-50.
EffectsofsupporttypetotheCOhydrogenationperformanceandactivationenergyofcobalt-basedcatalyticmaterials
XINGYu,LIUZhenxin,XUEYingying,WUDepeng,FANGShaoming
(HenanProvincialKeyLaboratoryofSurfaceandInterfaceScience,SchoolofMaterialsandChemicalEngineering,ZhengzhouUniversityofLightIndustry,Zhengzhou450002,China)
Underthereductiontemperatureof450-550 ℃,twotypesofcrystallinecobaltspeciescoexistwithinCo/ZnOcatalyticmaterial.Therearemanyliteratureaboutthereactionsofcobalt-basedcatalyticmaterialsinFischer-Tropschsynthesis(FTS).ComparisonoftheF-TcatalyticperformanceofcobaltmaterialsloadedoveraLewisbasicsupport(ZnO)aswellasaLewisacidicsupport(γ-Al2O3)wereconductedwithaspacevelocity(SV)of1 500mL/gcat·h,atotalpressureof2.0MPaandaH2toCOratioof2∶1.ItwasfoundthatsupportswithdifferentacidicorbasicpropertiesmaysignificantlyaffecttheCOhydrogenationactivity,productselectivitiesandapparentactivationenergyofcobalt-basedcatalyticmaterials.
cobalt;catalyticmaterial;compositematerial;support;COhydrogenation
1001-9731(2016)05-05073-05
國家自然科學基金資助項目(NSFCNo.U1204202,21571161);河南省科技計劃資助項目(124300510041)
2015-03-19
2015-08-10 通訊作者:邢宇,E-mail:yuxing@zzuli.edu.cn,方少明
邢宇(1976-),男,河南溫縣人,校聘教授,留美博士、博士后,從事多相催化、鋰電正極材料等方面的研究。
TQ426.64
A
10.3969/j.issn.1001-9731.2016.05.013
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
