二價金屬改性鐵酸鋅用于丁烯氧化脫氫的催化性能研究
2016-09-05 12:23:56唐曉麗寧國慶
山東化工 2016年2期
關鍵詞:催化劑
唐曉麗,寧國慶
(1. 中國石油大學(北京)重質油國家重點實驗室,北京 102249;2. 中國石油化工股份有限公司青島安全工程研究院,山東 青島 266071 )
科研與開發
二價金屬改性鐵酸鋅用于丁烯氧化脫氫的催化性能研究
唐曉麗1,2,寧國慶1*
(1. 中國石油大學(北京)重質油國家重點實驗室,北京 102249;2. 中國石油化工股份有限公司青島安全工程研究院,山東 青島 266071 )
采用共沉淀法向鐵酸鋅催化劑中添加不同二價金屬(Mg、Ni、Co、Cu和Ca),并以1-丁烯為原料,對上述改性催化劑的丁烯氧化脫氫性能進行了考察。加入Mg以后,鐵酸鋅催化劑催化效果顯著提高,在最佳鋅鎂比3:1的條件下,獲得了1-丁烯轉化率90.3%,丁二烯選擇性92.7%和丁二烯收率83.8%。比表面積和孔分布測試、X射線衍射分析和氨氣程序升溫脫附測試表明,鐵酸鋅鎂催化劑具有較高催化效果的原因可歸結為:鐵酸鋅鎂催化劑較鐵酸鋅催化劑具有更高的比表面積、較小的平均孔徑、較低的結晶度以及較強的表面酸量。
催化劑;部分氧化;脫氫;丁烯;丁二烯;鐵酸鋅
丁二烯是一種重要的化工產品原料,在石油化工烯烴原料中的地位僅次于乙烯和丙烯。它是合成橡膠的主要原料,可以生產順丁橡膠、丁苯橡膠、丁腈橡膠、氯丁橡膠、丙烯腈-丁二烯-苯乙烯、苯乙烯-丁二烯-苯乙烯彈性體等多種橡膠產品,還可以用來生產尼龍66、己二腈、己二胺等多種有機化工產品以及作為粘結劑和汽油添加劑等[1]。而丁烯氧化脫氫技術是制取丁二烯的重要方式之一[2-3]。丁烯氧化脫氫催化劑主要有鉬酸鉍系列[4-9]、混合氧化物Fe-Sb系列、鐵系尖晶石系列[10-16]以及磷酸鎳鈣系列等,此外還有采用碳材料[17]和負載型催化劑(例如Pd/SiO2[18]、Fe2O3/Al2O3和Fe2O3/SiO2[19])的報道。鉬酸鉍系列是較早采用的催化劑,雖然取得了較好的催化效果,例如Park等[8]報道的BiMoFex催化劑可獲得丁烯轉化率71%,丁二烯選擇性和產率分別為79%和56%,但同時存在著以下缺點:鉬組分流失嚴重,催化劑選擇性不高,副產物中含氧化合物生產量較高,而且會產生環境污染[20]。而鐵系尖晶石系列催化劑作為新一代催化劑,具有成本低、催化活性高(例如ZnFe2O4催化劑可獲得丁二烯產率67.8%[11])、無毒等優點,因而得到廣泛研究和生產應用[21]。鐵系尖晶石型催化劑的通式為MeⅡFe2O4,其中Me可以為Zn、Mg、Ni、Co等。
已有文獻報導采用不同二價金屬離子的鐵酸鹽催化劑,包括鐵酸鋅、鐵酸錳、鐵酸鎳、鐵酸鈷等鐵酸鹽催化劑[10-11,22-23]。例如,Lee等人[11]對鐵系尖晶石型催化劑中不同二價金屬的催化效果進行了考察,結果發現丁烯轉化率、丁二烯的選擇性和收率按下面次序排列ZnFe2O4>MgFe2O4>MnFe2O4>NiFe2O4>CoFe2O4>CuFe2O4,以鐵酸鋅為最佳。進一步,Lee等[13]針對鐵酸鋅催化劑研究了引入三價金屬組分的影響,結果表明丁二烯的產率按下面次序排列ZnFeFeO4(ZnFe2O4)>ZnAlFeO4>ZnCrFeO4>ZnMnFeO4>ZnCoFeO4,仍然以鐵酸鋅為最佳。盡管已有較多工作開展,目前采用兩種二價金屬的鐵系尖晶石型催化劑的研究仍未見報道。本文在鐵酸鋅催化劑的基礎上,分別向其中添加二價金屬Mg、Ni、Co、Cu和Ca,對它們的丁烯氧化脫氫催化效果進行了系統研究。研究結果表明,添加Mg的催化劑Zn0.75Mg0.25Fe2O4表現出比ZnFe2O4更高的催化性能,這為進一步提高鐵系尖晶石系列催化劑性能提供了新的途徑。
1 實驗部分
1.1 催化劑制備
取一定量的硝酸鐵、硝酸鋅及添加二價金屬對應的硝酸鹽溶解在蒸餾水中,然后用25%的氨水沉淀,沉淀終點pH約8.5~9,沉淀結束后在70~80 ℃下熱老化30 min,靜置1 h后過濾,濾餅在100 ℃下干燥12 h,然后再650 ℃下煅燒10 h,自然冷卻后破碎過篩,取20~60目用于活性評價。
1.2 表征
通過高分辨透射電鏡(HRTEM)(荷蘭FEI公司Tecnai G2 F20 Super-twin)觀察鐵系尖晶石型催化劑的晶格結構;通過比表面積測定儀(美國Micromeritic公司ASAP2020型吸附儀)測定催化劑的比表面積;通過氨氣程序升溫脫附(NH3-TPD)測定催化劑的表面酸量,具體方法為:取0.5 g催化劑于石英管中,在500 ℃下通入流量為20 mL/min的N2處理1 h以脫除樣品表面吸附的雜質,然后等到溫度降到110 ℃后通入NH3,維持0.5 h以保證樣品吸附飽和,然后再通入N2吹掃2 h以除去樣品表面發生物理吸附而吸附的NH3,待檢測器基線走平后,再以10 ℃/min的速率升溫至600 ℃,使樣品表面發生化學吸附而吸附的NH3脫附下來,采用TCD檢測器檢測NH3信號,并將脫附出來的NH3通入稀鹽酸溶液中進行吸收,然后用同濃度的NaOH溶液進行滴定,從而計算出樣品的總算量。
1.3 催化反應
鐵系尖晶石型催化劑上1-丁烯氧化脫氫反應的性能評價在內徑10 mm的石英管反應器的下行固定床微反裝置上進行。除特殊說明,一般評價過程為:將2.5 g催化劑裝入石英管反應器的中部,催化劑上部和下部的空間裝填石英棉。然后催化劑在溫度400 ℃,氧氣和氮氣流量分別為8 mL/min和32 mL/min的條件下預處理1 h,然后在反應溫度340 ℃,Vbutene:VO2:VH2O(g)=1:1:20,總體積流量Ft=250 mL/min的條件下進行催化劑評價。反應后的物料通過裝有氫火焰檢測器和熱導檢測器的SP-6890型氣相色譜儀進行在線監測,采用標準氣進行定性定量。1-丁烯轉化率和丁二烯選擇性分別采用(1)式和(2)式計算而得,丁二烯收率為二者乘積,見(3)式。
(1)
(2)
丁二烯收率=丁烯轉化率×丁二烯選擇性
(3)
2 結果與討論
2.1 催化劑表征
通過氮氣吸附-脫附等溫曲線可以得到催化劑的表面性質、空隙結構等信息。由圖1a中可以看出鐵酸鋅和鐵酸鋅鎂的N2吸附等溫曲線均屬于IUPAC分類中的Ⅴ型,H1型滯后環。相比于鐵酸鋅催化劑,鐵酸鋅鎂催化劑滯后環左移,最大孔徑分布左移。采用BET(Brunnauer-Emmett-Teller)方法計算得到,鐵酸鋅的比表面積為6.2 m2g-1,鐵酸鋅鎂催化劑的比表面積為15.2 m2g-1。由BJH法得到,鐵酸鋅催化劑的平均孔徑為44.5 nm,鐵酸鋅鎂催化劑的平均孔徑為34.9 nm(圖1b),這與N2等溫吸附脫附曲線中鐵酸鋅鎂的滯后環比鐵酸鋅的滯后環向小孔徑方向偏移是一致的。因此,我們推斷,鐵酸鋅鎂催化劑比鐵酸鋅催化劑具有更小的孔徑、更大的比表面積,從而可以增加催化劑與反應物丁烯的接觸面積。
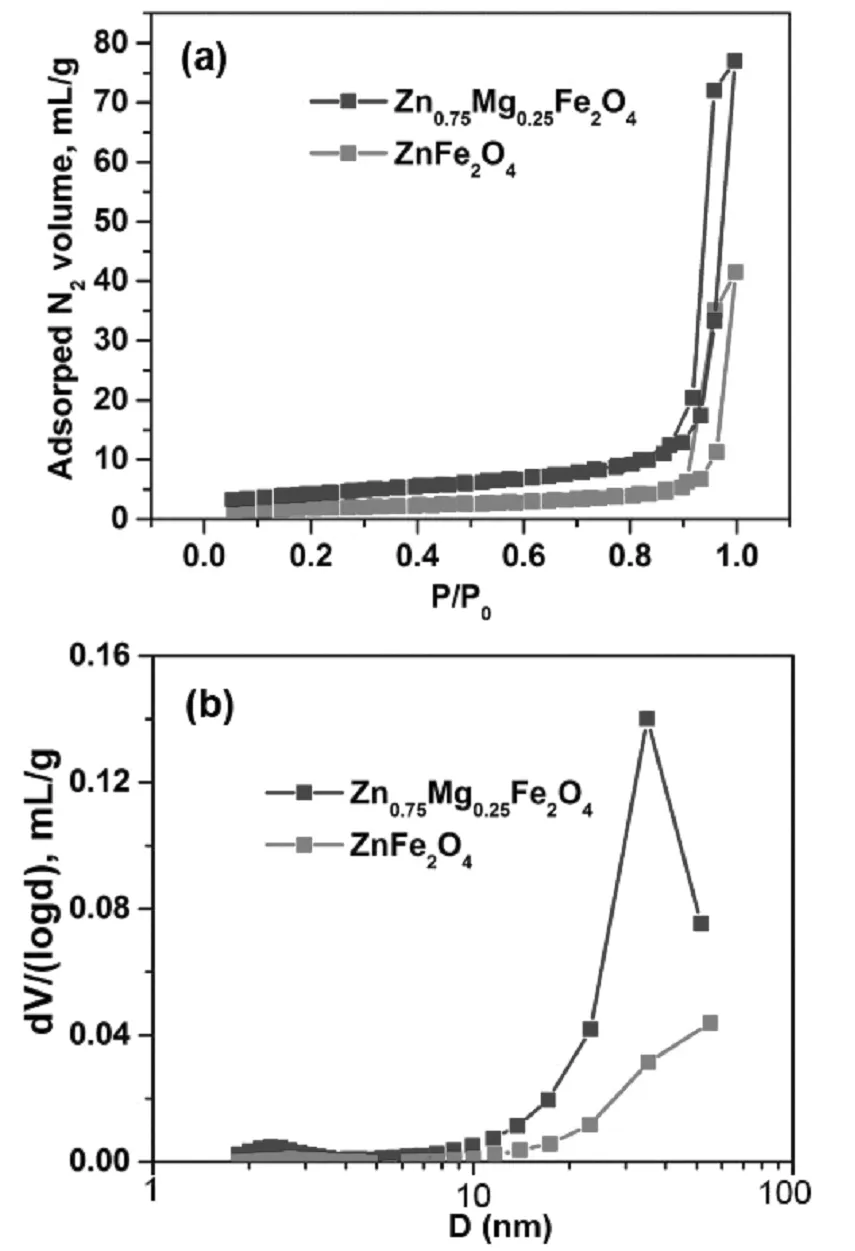
圖1 鐵酸鋅和鐵酸鋅鎂的氮吸附脫
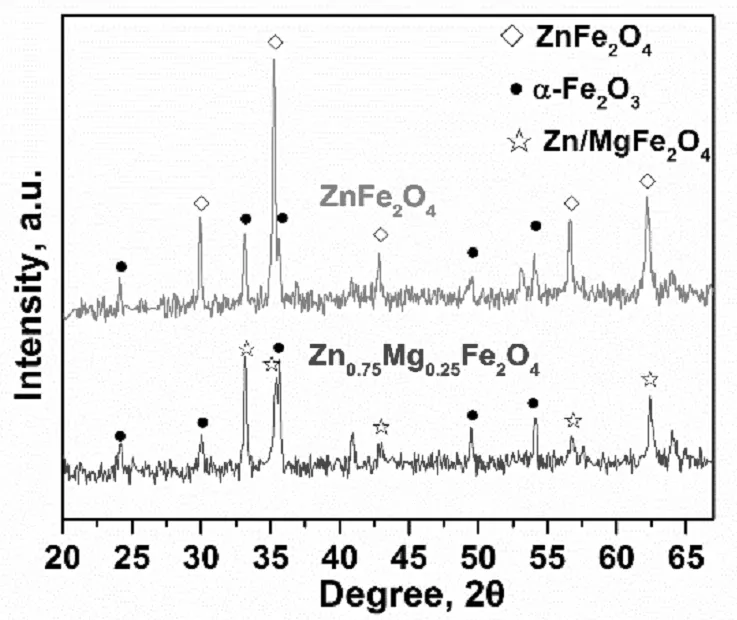
圖2 鐵酸鋅和鐵酸鋅鎂的XRD譜圖
為了考察鐵酸鋅和鐵酸鋅鎂催化劑的晶相結構,對它們進行了X 射線粉末衍射(XRD)表征,如圖2所示。ZnFe2O4的標準譜圖特征峰包含30.0°、35.3°、42.8°、56.6°和62.2°五處,其對應的晶面分別為[220]、[311]、[400]、[511]和[440];α-Fe2O3的標準譜圖主要特征峰包含24.1°、33.1°、35.6°、49.5°和54.1°五處,對應的晶面分別為[012]、[104]、[110]、[024]、[116]。從圖中可以看出,鐵酸鋅的XRD譜圖中包含了鐵酸鋅和α-Fe2O3兩種晶相。而由于MgFe2O4催化劑的標準譜圖特征峰同樣包含30.1°、35.4、43.1°、57.0°和62.5°五處,晶面分別為[220]、[311]、[400]、[511]和[440],這與ZnFe2O4的X射線衍射角以及晶面都是基本一致的,說明這兩種物相具有類似的晶相結構。因此,在鐵酸鋅鎂催化劑中含有α-Fe2O3、ZnFe2O4和MgFe2O4三種晶相。對比鐵酸鋅催化劑的XRD譜圖可以看到,由于引入Mg2+鐵酸鋅鎂催化劑的Zn/MgFe2O4相的衍射峰強度明顯降低,說明Zn0.75Mg0.25Fe2O4晶相結晶度較單純的ZnFe2O4晶相結晶度降低。我們知道,鐵酸鹽催化劑作為1-丁烯氧化脫氫催化劑時,氧將Fe2+氧化為Fe3+是恢復催化劑活性的重要一環,而如果結晶度過高,氧在晶格間的移動性會受到嚴重抑制,從而導致催化劑的活性降低。
2.2 不同金屬添加劑對氧化脫氫反應催化性能的影響
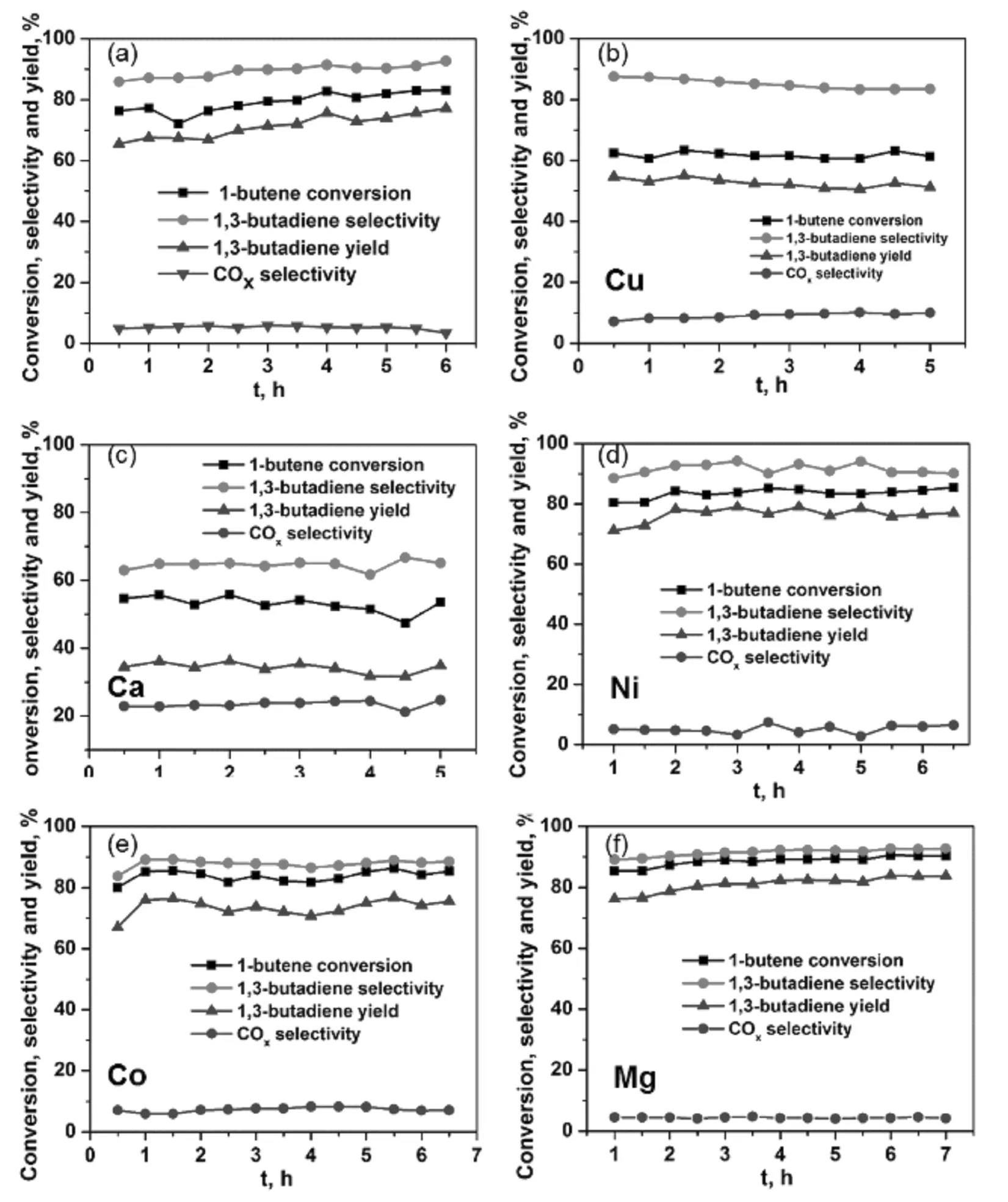
圖3 純鐵酸鋅催化劑(a)和二價金屬添加的鐵酸鋅催化劑(b-f)的1-丁烯氧化脫氫催化性能。
將幾種常見的二價金屬加入到鐵酸鋅(ZnFe2O4)催化劑中,保持物質的量比Fe/Zn/Me=2.3/0.75/0.25(Me=Ca、Co、Cu、Mg、Ni),其他制備條件與鐵酸鋅催化劑的制備條件相同,考察添加二價金屬對鐵酸鋅催化劑催化效果的影響。作為對比,我們也制備了純的鐵酸鋅催化劑,Fe/Zn=2.3:1。反應條件為溫度340 ℃,氧烯比1.0,水烯比20,催化劑2.5 g。如圖3a所示,采用純鐵酸鋅作為催化劑,1-丁烯轉化率可達83.1%,丁二烯選擇性可達92.7%,丁二烯收率最高達77.1%。從圖3b中可以看到引入Cu以后,而1-丁烯的轉化率則有大幅度降低,使得丁二烯的收率僅為55%左右;Ca的引入對鐵酸鋅催化劑的催化效果起抑制作用(圖3c),無論是1-丁烯的轉化率還是丁二烯的選擇性均大幅度降低,而碳氧化物的選擇性則大幅上升,最高達24.7%,丁二烯的收率僅為35%左右;而引入金屬Ni以后(圖3d),可以看到,我們得到了較高的丁二烯選擇性,基本維持在90%以上,而1-丁烯的轉化率在85%左右,因此取得了和單純鐵酸鋅催化劑相當的催化效果;引入Co以后的催化效果(圖3e)比引入Ni的效果略差;當向鐵酸鋅催化劑中加入Mg2+以后,如圖3f所示,可以看到三條曲線整體呈攀升態勢,并且反應3 h以后,無論是1-丁烯轉化率,還是丁二烯的選擇性和收率均比鐵酸鋅催化劑有所提高,反應7 h時,1-丁烯的轉化率以及丁二烯的選擇性和收率分別為90.3%,92.7%和83.8%。我們獲得的丁二烯收率(83.8%)略高于Lee等報道的加入三價金屬Fe改性的ZnMeIIIFeO4獲得的收率(79.7%)[13],顯著高于BiFe0.65NixMo氧化物催化劑獲得的丁二烯收率(72%)[9]和碳納米催化劑獲得的丁二烯收率(43%)[17]。

圖4 不同金屬添加劑對丁二烯的影響(a)和
圖4a對上述幾種催化劑的丁二烯收率進行了對比。從圖中可以看到添加Mg以后得到了最大的丁二烯收率,其次分別是Ni、Co、Cu和Ca。對Mg改性的鐵酸鋅催化劑,保持Fe的摩爾量以及Zn和Mg的總摩爾數不變,調變Zn和Mg的摩爾比,我們進一步考察了不同的鋅鎂比對催化反應結果的影響。從圖4b中可以看到,當鋅含量比較高的時候(Zn/Mg=5/1和3/1),催化劑的催化效果較純鐵酸鋅催化劑(Zn/Mg=1/0)有所提高,當Zn/Mg為3:1時,催化劑的活性達到最大值,1-丁烯轉化率、丁二烯選擇性和收率分別達88.0%,91.5%和80.5%;而當鎂含量繼續增大到Zn/Mg=1/1時,1-丁烯轉化率,丁二烯的選擇性和收率則開始下降,并且要低于Zn/Mg=5/1時的催化效果;而當鐵酸鋅催化劑中的Zn完全被Mg代替(Zn/Mg=0/1)時,鐵酸鎂催化劑的催化效果要低于鐵酸鋅和鐵酸鋅鎂催化劑,1-丁烯的轉化率進一步降低到78.4%,丁二烯的選擇性變化不大,而丁二烯的收率僅為68.4%。
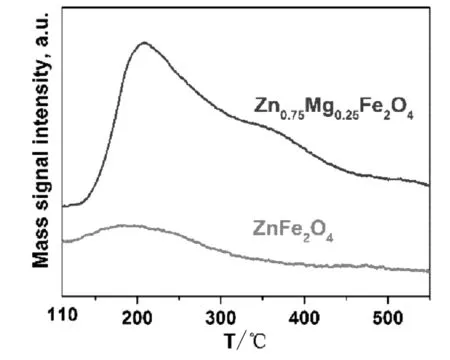
圖5 鐵酸鋅和鐵酸鋅鎂的NH3-TPD圖。
2.3 添加劑影響機理分析
NH3-TPD是表征固體材料表面酸性質的有效技術,通常用來測定催化劑或其載體的酸量、酸強度和酸強度分布等參數。其原理是采用分子尺寸小于被測固體材料孔徑的NH3作為探針分子,使其在被測固體材料的表面酸性位吸附至飽和,然后通過惰性氣體吹掃去除物理吸附的NH3,則材料表面剩余的為化學吸附的NH3,然后通過程序升溫脫除被吸附的NH3,并通過TCD記錄下NH3脫附過程的信號,得到該材料的NH3-TPD曲線。該脫附曲線可有效反映被測固體的酸量和酸強度分布。一般根據脫附峰溫度區間可把NH3-TPD曲線分為弱酸(<250 ℃)、中強酸(250~450 ℃)和強酸(>450 ℃)三類酸強度。由于催化劑的表面酸性質對其活性具有重要影響,所以我們考察了鐵酸鋅催化劑和鐵酸鋅鎂催化劑的催化活性,如圖5所示。從圖5中可以看出,鐵酸鋅催化劑和鐵酸鋅鎂催化劑都呈一個連續的酸強度分布。鐵酸鋅催化劑在190 ℃有一個面積較小的NH3脫附峰,而在250 ℃以后沒有明顯的峰形,說明鐵酸鋅催化劑的表面酸性位主要為弱酸,且酸量較小;而鐵酸鋅鎂的NH3-TPD曲線上可以看到,在210 ℃出現了一個較大的NH3脫附峰,脫附峰位置右移,并且在350 ℃附近出現了一個不明顯的凸包,說明鐵酸鋅鎂催化劑表面多的弱酸活性位和少量的中強酸活性位,酸強度較鐵酸鋅催化劑變大。由滴定法測定的鐵酸鋅和鐵酸鋅鎂催化劑的總酸量分別為88.9 μmol NH3/g催化劑和102.5 μmol NH3/g催化劑。
Lee等考察了鐵酸鋅沉淀的pH發現,當pH值為6~12時可以形成鐵酸鋅相,當沉淀pH值為6~10時,催化劑的活性較高,通過NH3-TPD研究發現,丁烯轉化率隨著催化劑表面酸度增大而增大,而當沉淀pH值=9時,催化劑的表面酸度最大[6]。另外,他們還利用硫酸銨溶液浸漬的方法硫化鐵酸鋅催化劑,結果發現硫化后的鐵酸鋅催化劑活性提高,這是由于硫化使鐵酸鋅催化劑的酸性位增多,而堿性位減少[5]。由此可見,催化劑的酸量越強,越有利于1-丁烯氧化脫氫反應。前面的測定結果顯示鐵酸鋅鎂催化劑表面酸強度比鐵酸鋅催化劑大,這可能是鎂改性的鐵酸鋅催化劑具有更高催化性能的原因之一。
3 結論
向鐵酸鋅催化劑中添加Cu、Mg、Ni、Ca和Co對其進行改性研究,結果發現,加入Mg以后,1-丁烯轉化率和丁二烯選擇性均有所提高,Ni和Co對反應結果影響不大,而加入Cu和Ca對1-丁烯的氧化脫氫反應時不利的。最佳的鋅鎂比為3/1。通過氮吸附脫附分析鐵酸鋅鎂催化劑具有更大的比表面積和更小的孔徑,通過XRD譜圖分析鐵酸鋅催化劑中包含ZnFe2O4和α-Fe2O3兩種晶相,鐵酸鋅鎂催化劑中包含ZnFe2O4、MgFe2O4和α-Fe2O3三種晶相,但是鐵酸鋅鎂催化劑較鐵酸鋅催化劑結晶度降低。NH3-TPD結果表明,鐵酸鋅催化劑和鐵酸鋅鎂催化劑表面酸性均呈弱酸性,但是鐵酸鋅鎂催化劑的酸性更強。因此,我們推測,較大的比表面積,較小的結晶度以及較強的表面酸性更有利于1-丁烯的氧化脫氫反應。
[1] 崔小明. 丁二烯生產技術進展及國內外市場分析[J].精細石油化工進展, 2010(11):34-41.
[2] 黃水源,汪展文,金 涌,等. 氣固固循環流化反應器丁烯氧化脫氫制丁二烯[J]. 化工學報, 1997, 48(06):673-679.
[3] 張吉瑞, 王家強,王玉慶. B-02催化劑上丁烯氧化脫氫動力學研究[J]. 化工學報, 1991, 42(5):617-624.
[4] Ruckenstein E, Krishnan R, Rai K N. Oxygen depletion of oxide catalysts[J].Journal of Catalysis, 1976, 45(2):270-273.
[5] Jung J C, Kim H, Choi A S, et al. Effect of pH in the preparation of γ-Bi2MoO6 for oxidative dehydrogenation of n-butene to 1,3-butadiene: Correlation between catalytic performance and oxygen mobility of γ-Bi2MoO6[J].Catalysis Communications, 2007, 8(3):625-628.
[6] Jung J C, Lee H, Song I K. Effect of preparation method of Co9Fe3Bi1Mo12O51on the catalytic performance in the oxidative dehydrogenation of n-butene to 1,3-butadiene-comparison between Co-precipitation method and citric acid-derived sol-gel method[J]. Catalysis Letters, 2009, 128(1/2):243-247.
[7] Jung J C, Lee H, Seo J G, et al. Oxidative dehydrogenation of n-butene to 1,3-butadiene over multicomponent bismuth molybdate ((M9Fe3Bi1Mo12O51)-Fe-II) catalysts: Effect of divalent metal (M-II)[J].Catalysis Today, 2009, 141(3/4): 325-329.
[8] Park J H, Noh H, Park J W, et al. Effects of iron content on bismuth molybdate for the oxidative dehydrogenation of n-butenes to 1,3-butadiene[J].Applied Catalysis a-General, 2012, 431: 137-143.
[9] Park J H, Row K, Shin C H. Oxidative dehydrogenation of 1-butene to 1,3-butadiene over BiFe0.65NixMo oxide catalysts: Effect of nickel content[J].Catalysis Communications, 2013, 31: 76-80.
[10] Lee H, Jung J C, Kim H, et al. Effect of pH in the preparation of ZnFe2O4for oxidative dehydrogenation of n-butene to 1,3-butadiene: Correlation between catalytic performance and surface acidity of ZnFe2O4[J]. Catalysis Communications, 2008, 9(6): 1137-1142.
[11] Lee H, Jung J C, Kim H, et al. Effect of divalent metal component (Me(II)) on the catalytic performance of Me(II)Fe2O4catalysts in the oxidative dehydrogenation of n-butene to 1,3-butadiene[J]. Catalysis Letters, 2008, 124(3/4):364-368.
[12] Lee H, Jung J, Song I. Oxidative dehydrogenation of n-butene to 1,3-butadiene over sulfated ZnFe2O4Catalyst[J]. Catalysis Letters, 2009, 133(3/4): 321-327.
[13] Lee H, Jung J C, Kim H, et al. Oxidative dehydrogenation of n-butene to 1,3-butadiene over (ZnMeFeO4)-Fe-III catalysts: effect of trivalent metal (Me-III)[J]. Catalysis Letters, 2009, 131(3/4): 344-349.
[14] Lee H, Jung J C, Kim H, et al. Effect of Cs (x) H3-x PW12O40 addition on the catalytic performance of ZnFe2O4in the oxidative dehydrogenation of n-butene to 1,3-butadiene[J]. Korean Journal of Chemical Engineering, 2009, 26(4):994-998.
[15] Chung Y M, Kwon Y T, Kim T J, et al. Factors affect on the reaction performance of the oxidative dehydrogenation of n-butene to 1,3-butadiene over Zn-Ferrite catalysts[J]. Catalysis Letters, 2009, 130(3/4): 417-423.
[16] Chung Y M, Kwon Y T, Kim T J, et al. Prevention of catalyst deactivation in the oxidative dehydrogenation of n-butene to 1,3-butadiene over Zn-Ferrite catalysts[J]. Catalysis Letters, 2009, 131(3/4): 579-586.
[17] Liu X, Su D S, Schloegl R. Oxidative dehydrogenation of 1-butene to butadiene over carbon nanotube catalysts[J]. Carbon, 2008, 46(3):547-549.
[18] Furukawa S, Endo M, Komatsu T. Bifunctional catalytic system effective for oxidative dehydrogenation of 1-butene and n-butane using Pd-based intermetallic compounds[J]. Acs Catalysis, 2014, 4(10): 3533-3542.
[19] Yan W, Kouk Q Y, Luo J, et al. Catalytic oxidative dehydrogenation of 1-butene to 1,3-butadiene using CO2[J]. Catalysis Communications, 2014, 46: 208-212.
[20] 李親華. 我國丁烯氧化脫氫制丁二烯技術進展[J].石油化工, 1990,19 (03): 183-190.
[21] 楊鳳琨,陳獻誠,劉俊聲,等. 丁烯氧化脫氫R系列鐵系催化劑的研究 [J]. 石油化工, 1997 (10):3-7.
[22] Gibson M A, Hightower J W. Oxidative dehydrogenation of butenes over magnesium ferrite: Catalyst deactivation studies[J]. Journal of Catalysis, 1976, 41(3): 431-439.
[23] Cares W R, Hightower J W. Ferrite spinels as catalysts in the oxidative dehydrogenation of butenes[J]. Journal of Catalysis, 1971, 23(2): 193-203.
(本文文獻格式:唐曉麗,寧國慶.二價金屬改性鐵酸鋅用于丁烯氧化脫氫的催化性能研究[J].山東化工,2016,45(02):1-5.)
Effect of Divalent Metal Addition into ZnFe2O4Catalysts on the Catalytic Performance for Oxidative Dehydrogenation of 1-Butene
Tang Xiaoli1,2, Ning Guoqing1
(1. State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing 102249,China;2.Research Institute of Safety Engineering, SINOPEC, Qingdao 266071,China)
ZnFe2O4catalysts doped by divalent metals (Mg, Ni, Co, Cu and Ca) are prepared by a co-precipitation method, to catalyze the oxidative dehydrogenation of 1-butene to 1,3-butadiene. The Mg-doped ZnFe2O4catalyst exhibits the best catalytic performance among all the doped catalysts. At the optimized Zn to Mg atom ratio of 3:1, the 1-butene conversion of 90.3%, the 1,3-butadiene selectivity of 92.7% and the 1,3-butadiene yield of 83.8% are obtained. The enhanced catalytic performance of the Mg-doped ZnFe2O4can be attributed to its higher specific surface area, smaller pore diameter, smaller degree of crystallinity and stronger surface acidity as compared to the undoped ZnFe2O4catalyst, as revealed by nitrogen adsorption-desorption, X-ray diffraction and NH3-temperature programmed desorption characterizations.
catalyst; partial oxidation; dehydrogenation; butene; butadiene; zinc ferrite
2015-12-02
博士點基金(20110007120004)
寧國慶(1979—),山東泰安人,副研究員,主要從事碳材料和發應工程研究。
O643.36
A
1008-021X(2016)02-0001-05
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
