Ag-La系二元化合物結構與電子性能的第一性原理研究
2016-09-13 08:33:52高恩強張照超阮海光黃福祥陳志謙王蘭蘭
重慶理工大學學報(自然科學) 2016年8期
關鍵詞:結構
高恩強,張照超,阮海光,黃福祥,陳志謙,王蘭蘭
(1.重慶理工大學 材料科學與工程學院,重慶 400054;2.西南大學 材料與能源學部,重慶 400715)
?
Ag-La系二元化合物結構與電子性能的第一性原理研究
高恩強1,張照超1,阮海光1,黃福祥1,陳志謙2,王蘭蘭1
(1.重慶理工大學 材料科學與工程學院,重慶400054;2.西南大學 材料與能源學部,重慶400715)
通過第一性原理方法系統地研究了Ag-La系四種二元化合物的相結構穩定性和電子結構,包括B2-LaAg、LaAg2、La14Ag51和α-LaAg5。結構優化后的平衡態晶體參數及質量密度與實驗值相符。結合能表明,隨La濃度的增加,化合物的鍵合強度和穩定性提高。Ag-La系4種二元化合物的生成焓分別為-21.7,-26.8,-22.9,-18.1 kJ/mol,與實驗值或CALPHAD的理論值相符。電子結構表明這些化合物是導體,其原子間的價鍵性質是由金屬鍵、離子鍵和共價鍵構成,其中:離子鍵是由Ag原子從La原子中得到電子形成的,共價鍵是由Ag s-p及Ag p-La d雜化構成的。并且這些化合物的共價鍵和離子鍵隨著La濃度的增加而增強,使得Ag-La化合物穩定性提高。
Ag-La系;稀土相;電子結構;第一性原理
直到目前為止,對Ag-La系化合物的物理化學性質研究,無論是實驗上還理論上還是很匱乏。另一方面,為了更深入了解Ag-La系二元化合物則需要更系統的數據。本文將采用第一性原理計算方法,系統地研究Ag-La系二元化合物的晶體結構和電子結構的關系,包括B2-LaAg、LaAg2、La14Ag51和α-LaAg5。
1 計算細節
本文的第一性原理計算由基于密度泛函理論的CASTEP[20-21]執行,使用Vanderbit類型的超軟贗勢[22]和平面波函數的基組[23]。電子交換-關聯能泛函為廣義梯度近似(GGA)perdew-burke-ernzerhof(PBE)形式[24]。總能計算采用自洽迭代法(SCF),SCF收斂差值為5×10-7eV/atom。幾何優化的截斷能設置為350 eV,第一布里淵區k點取樣[25]為0.04 eV/10-10m,采用broyden-flecher-goldfarb-shanno(BFGS)方法[26]尋找最穩定結構。所有晶胞在自洽計算時,體系總能量的收斂值取5×10-6eV/atom,每個原子上的力低于0.01 eV/10-10m,公差偏移小于5×10-14m,應力偏差小于0.02 GPa。價電子排布為:Ag4d105s1,La 5s25p65d16s2。
2 結果與分析
2.1晶體結構
圖1為Ag-La系二元化合物的晶體結構。為研究這些化合物的基態性質,本文計算了Ag-La系的二元化合物平衡態的晶體參數。表1給出了這4種化合物幾何優化后的晶體結構及相應的實驗值。在低溫六方MgZn2型α-LaAg5相中,在Wyckoff4f位置包含2個Ag原子和2個La原子,因其相對位置不同,可分為3種結構:LaAg5-f1f2,-f1f3及-f2f3,其中:LaAg5-f1f2的晶格常數c與實驗值相差較大,為7.9%;LaAg5-f2f3的晶格常數a也有較大誤差,達2.4%。本文的其他計算值和實驗值很相符,誤差小于2%,即理論計算的誤差在可接受范圍內。表1顯示,本文的第一計算方法所給出的可靠精確值比實驗值稍大。這種相對誤差可能與本文計算時忽略溫度因素和所選取的交換-關聯泛函GGA-PBE有關。

圖1 Ag-La系金屬間化合物的晶體結構

相空間群Pearson密度ρ/(g·cm-3)晶格常數a/10-10mB/10-10mc/10-10mLaAg5-f1f2P63/mmmhP128.63495.54549.795LaAg5-f1f3P63/mmmhP128.87685.63619.224LaAg5-f2f3P63/mmmhP128.74715.70029.15139.23885.5699.0775La14Ag51P6/MhP658.559613.13839.66288.9312.9559.525LaAg2ImmaoI127.80014.87247.49298.27178.1744.8257.3878.196B2-LaAgPm3mcP27.24433.83877.483.8147.3052
圖2描述了Ag-La系二元化合物的質量密度與La濃度間的關系,包含實驗值[14](Exp.)和理論值[27]。與實驗值相比,本文(GGA-PBE)所得的Ag-La 系化合物的質量密度偏低。正如圖2所示,純銀的密度(10.137 7 g/cm3)大于純鑭的密度(5.810 1 g/cm3),因此隨著La濃度的增加,Ag-La系化合物的質量密度隨La濃度的增加幾乎呈線性下降,線性擬合方程為:ρ=9.500 2-3.998 4xLa。與晶格參數一樣,LaAg5中的3種結構f1f3密度最大且與實驗值擬合的最好,其次是f2f3,最后是f1f2。

圖2 Ag-La系金屬間化合物的理論密度(GGA-PBE)、
2.2熱力學性能
為研究Ag-La系二元化合物的熱力學性能,本文計算了4個Ag-La系化合物及純Ag、La的基態總能以及Ag、La的孤立原子能量。對于確定的Ag-La系化合物LaaAgb,生成焓(Ef)的定義為LaaAgb總能與純Ag、La固態能線性組合的差值,結合能(Ecoh)定義為LaaAgb總能與Ag、La孤立原子能線性組合的差值。因而,Ag-La系化合物的形成焓與結合能的表達式為[28]:
(1)
(2)


表2 Ag-La二元系金屬間化合物的生成焓(Ef)和結合能(Ecoh)
注:生成焓和結合能皆為原子摩爾能。
事實上,Ef與Ecoh在預示結構穩定性上的偏差源于它們各自不同的物理意義。生成焓表示生成一摩爾原子化合物的焓變。眾所周知,自由能可以表示相的穩定性,在溫度為0 K時無熵貢獻,自由能等于生成焓。換句話說,生成焓可以用于確定化合物的相穩定性。另一方面,結合能是無相互作用的原子聚合的能量,它是衡量固態中原子結合鍵的強度,這與基態下結構穩定性相關。一般而言,在純組分形成金屬間化合物期間,其晶體結構、鍵的類型及強度都會發生變化。
Ag-La化合物的熱穩定性可以由計算的生成焓進行分析。從圖3可知,α-LaAg5,La14Ag51,LaAg2及B2-LaAg相是穩定相,其中α-LaAg5的3種結構中LaAg5-f1f3的值最小,并且其值及其他化學計量化合物的值與CALPHAD或實驗非常相符[16,18]。對于α-LaAg5金屬間化合物,f1f2,f1f3和f2f3的生成焓分別為-10.7,-18.1和-6.1 kJ/mol,這表明f1f3結構的熱力學穩定性最好,而f2f3結構的熱力學穩定性最差。La14Ag51,LaAg2及B2-LaAg的計算值分別為-22.9,-26.8和-21.7 kJ/mol。這些值表明Ag-La系金屬間化合物中的Ag與La原子間的化學相互作用并不是很強,但負值表明這些化合物是熱力學穩定的。
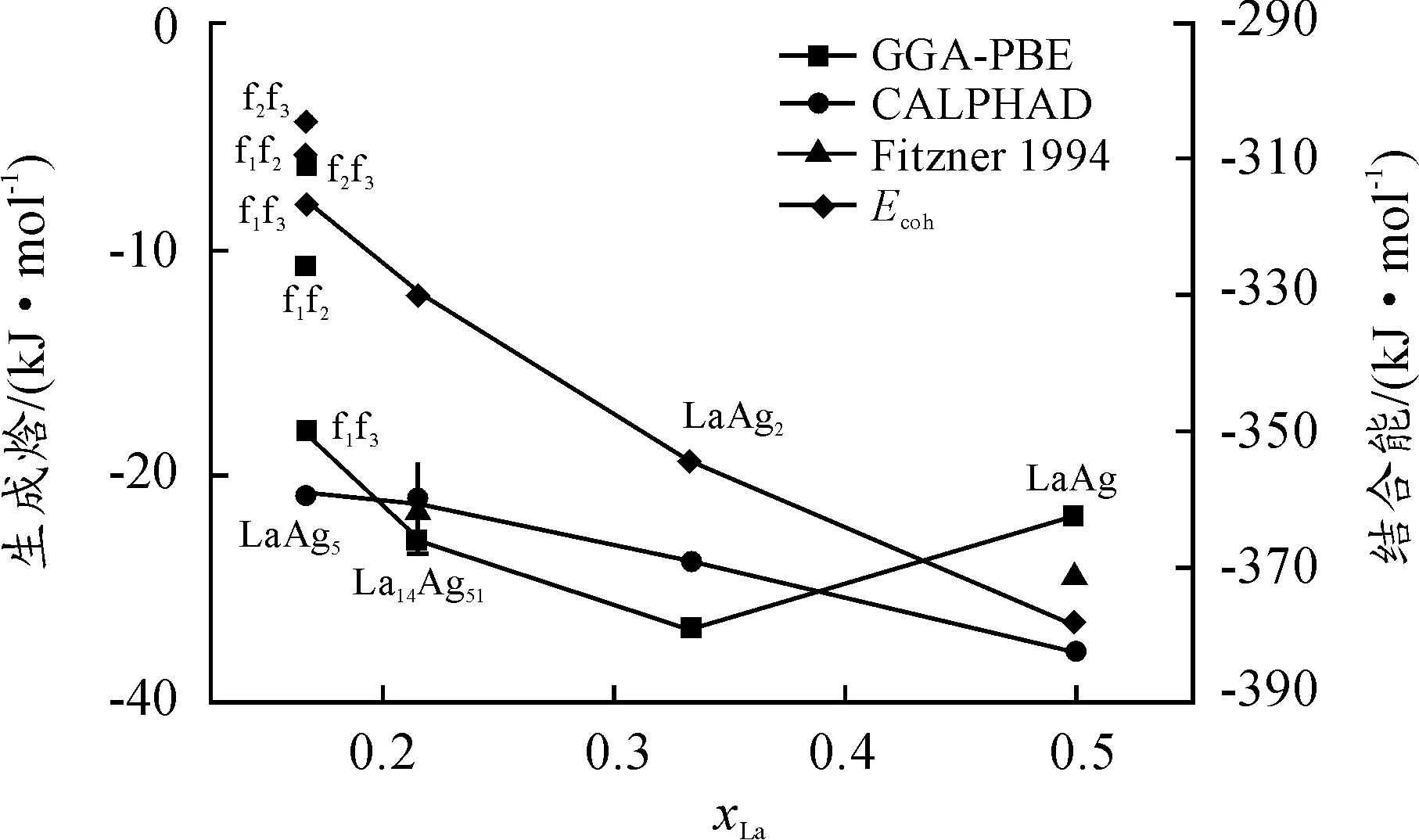
圖3 Ag-La系二元金屬間化合物的本文計算(GGA-PBE)、
結合能(Ecoh)是衡量材料穩定性的另一個重要平衡熱力學參數。根據式(2),表2和圖3總結了Ag-La系金屬間化合物的結合能。圖2表明Ag-La系二元化合物的結合能負值隨La濃度的增加而增大。這結果說明這些化合物的穩定性隨La濃度的增加而提高,這是因為對于確定的金屬間化合物的穩定性可以通過結合能反映,負值越小反映穩定性越高[30]。同樣,通過結合能也可以反映金屬間化合物中的Ag、La原子間的化學間相互作用的強度。
2.3電子結構
為了得到精確的Ag-La系二元金屬間化合物電子性能,本文依次從電子態密度(DOS)以及布居分析等方面詳細描述了化合物中的電子結構,如圖4和表3所示。
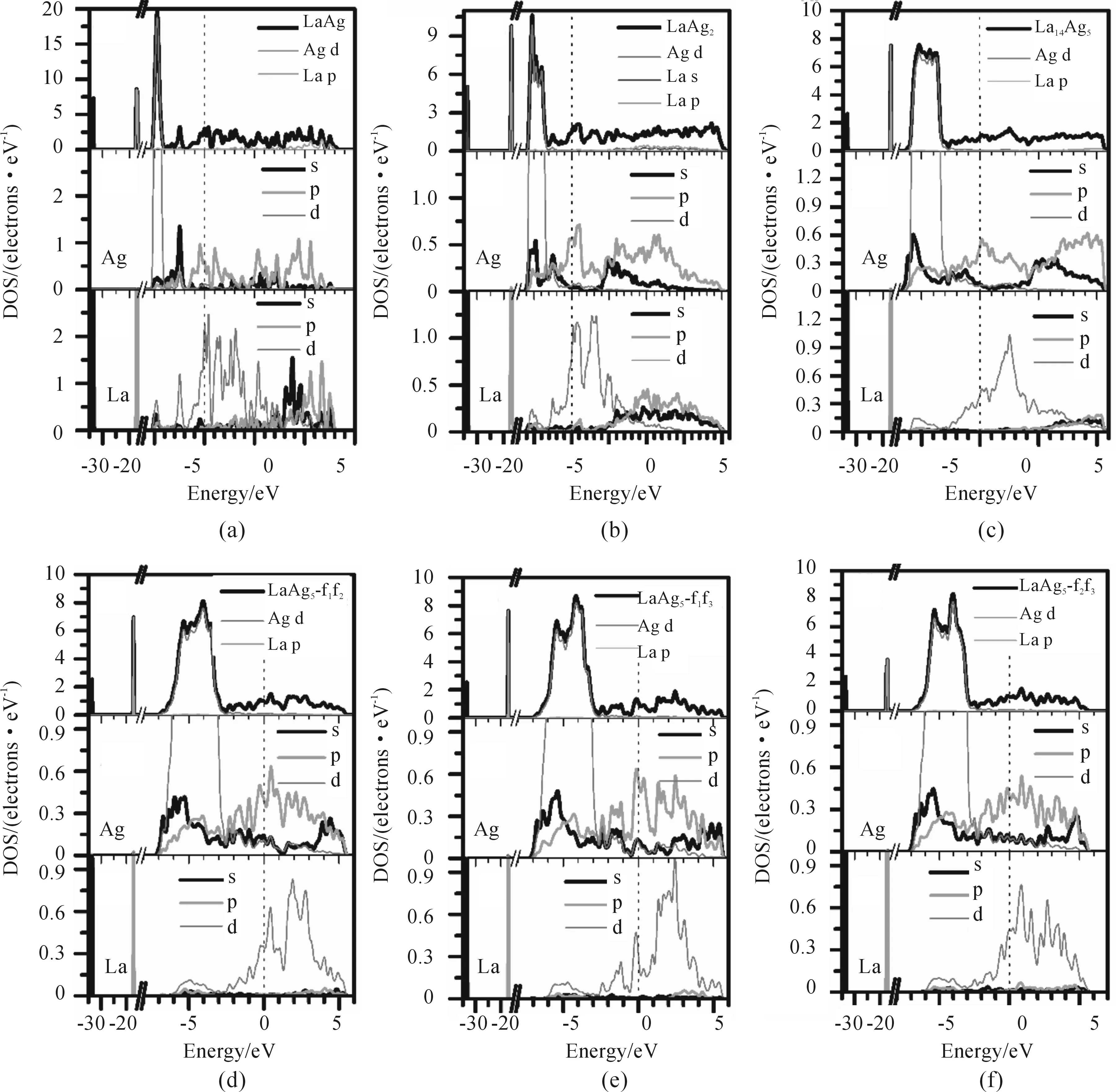
圖4 Ag-La系二元金屬間化合物的總態密度和分態密度,Fermi能級位于虛線能級為零處

相原子spdTotalCharge(e)LaAg5-f1f2Ag0.7920.8689.77311.433-0.433La1.0361.818.84+2.16LaAg5-f1f3Ag0.7840.8869.76811.438-0.438La1.035.871.898.79+2.21LaAg5-f2f3Ag0.7490.8719.77411.394-0.394La1.225.891.959.05+1.95

續表(表3)
在統計和固態物理學中,體系的態密度(DOS)用于描述每個能級處的可用于被占的電子態數目。某能級處的DOS越大意味著該處可用于電子占據的態越多。圖4為Ag-La化合物的總態和分態密度,其中Feimi能級被設定為0的參考能級。從圖4(a)~(f)可知,Ag-La系二元化合物在Fermi能級處不為0,這些化合物均為導體,具有金屬性質,其主要由La d和Ag p軌道構成。在低能級范圍(-35~-15 eV),總態密度主要歸因于La s和La p態。從LaAg到LaAg5,隨著Ag濃度的增加,Ag d的能級寬度也逐漸增加(-8~-2 eV),這種變化使鍵強產生差異,并且可能導致更高的力學模量。在-2 eV到Feimi能級之間部分主要由La d和Ag p構成,表明La d和Ag p對這些化合物的金屬行為起重要作用。在Feimi能級以上區域,態密度相對較低且平坦,這是因為Ag的最外層d軌道已被完全占據。微弱的低能級Ag s-p 成鍵雜化和高能級Ag p-La d成鍵或反鍵雜化說明Ag-La系二元金屬間化合物中存在共價鍵。對于LaAg5(圖4(d)~(f)),軌道雜化強度順序為f1f3>f1f2>f2f3,這可以與結構穩定性有關。
本文分析了Ag-La化合物原子的電荷布居分布,如表3所示。負號表示得電子,正號表示失電子。很明顯,Ag原子得到電子而La原子失去電子。Ag原子從La原子中得到電子,這可能是因為Ag與La原子間的電負性相差較大(χAg(1.93)>χLa(1.10)),引起La原子外層失去電子,從而Ag-La間的價鍵為離子鍵。隨著La濃度的增加,電子轉移量逐漸增加,離子性增強,其中α-LaAg5化合物中LaAg5-f1f3的電子轉移量最大,導致在這些晶體結構中LaAg5-f1f3的韌性受影響的程度最大。對比圖4分態密度,La s主要分成2部分:位于能級最低處以及Fermi能級以上。隨Ag的濃度增加,從LaAg2開始最低能級處的La s逐漸加大失電子量。由表3可知,Ag-La系化合物中La d和Ag p軌道從La s軌道得到電子,隨著La濃度的增加,Ags、p態電子數增加,表明Ag s-p雜化加強,同時La d態電子變化不明顯,Ag p-La d雜化也升高。金屬鍵可以認為是同種原子之間的電荷轉移,因而不同化合物價鍵的金屬性差異不是很大。另外,被廣泛認同的是,布居分析得到的原子電荷的絕對值大小沒有物理意義,因為布居分析對所計算的原子基組非常敏感。但是,相對的電子轉移可以用于定性描述體系變化。基于以上電子結構分析,得出Ag-La系化合物的化學鍵結構由金屬鍵、離子鍵和共價鍵構成。
3 結論
Ag-La系金屬間化合物B2-LaAg、LaAg2、La14Ag51及α-LaAg5的生成焓和結合能均為負值,為穩定相,并隨著La濃度的增加,結合能增加,穩定性升高,其中LaAg5的三種結構中LaAg5-f1f3最穩定。電子結構表明:Ag-La系化合物的價鍵包含了金屬性、離子性和共價性,其中離子性和共價性隨化合物的La、Ag原子計量比的增加而增加,進而導致化合物的原子間結合力提升,穩定性增加。共價性主要是由Ag s-p及Ag p-La d雜化構成,離子性是由于Ag、La原子間的電負性差異引起電荷轉移形成的。相鄰Ag原子間存在d態電子局域以及Ag s-p低能級弱雜化,相鄰Ag-La原子間的價鍵具有離子性和微弱的Ag p-La d雜化,而相鄰La原子間因正電性相斥僅存在金屬性。
[1]BERGER G,WEISS A.Ternary intermetallic phases with Heusler-phase-type structure in the system Ag-Mg-RE(RE≡La,Ce,Pr,Nd,Sm)[J].Journal of the Less Common Metals,1988,142:109-121.
[2]陳志軍.稀土對銀銅鎳合金組織及性能的影響[D].哈爾濱:哈爾濱工業大學,2010.
[3]毛利權,韓吉慶,趙濤,等.微量稀土Ce對Ag的力學性能的影響[J].鑄造技術,2010(12):1592-1595.
[4]HUANG F X,LI M,YING P,et al.Effect of trace cerium on the as-cast microstructure of Ag-Cu-Ni alloy[C]//proceedings of the Materials Science Forum.Trans Tech Publ.USA:[s.n.],2011.
[5]MA X,LI L F,ZHANG Z H,et al.Effects of rare earth La on microstructure and properties of Ag21Cu25Sn alloy ribbon prepared by melt spinning[J].Materials & Design,2015,83:1-5.
[6]PU C Y,ZHOU D W,SONG Y L,et al.Phase transition and thermodynamic properties of YAg alloy from first-principles calculations[J].Computational Materials Science,2015,102:21-26.
[7]TAMBORNINO F,SAPPL J,HOCH C.The Gd14Ag51structure type and its relation to some complex amalgam structures[J].Journal of Alloys and Compounds,2015,618:326-335.
[8]GAO E Q,HUANG F X,CHEN Z Q,et al.First-principles investigation of mechanical and electronic properties of LaAg5Laves phase under pressure[J].Journal of Rare Earths,2016,34(1):105-112.
[9]徐濤,胡小武,江雄心.基板稀土微合金化對Sn3Ag0.5Cu/Cu釬焊界面反應的影響[J].電子元件與材料,2016(2):65-69.
[10]敬小龍,鄧瑩,陳慧,等.稀土發光材料的第一性原理計算[J].功能材料,2016(2):02015-02019.
[11]VOGEL R,KLOSE H.On Ternary Systems with Two Rare Earths:Silver-Cerium-Lanthanum[J].Z Metallkd,1954,45:670-670.
[12]CANNERI G.Le leghe del lantanio[J].Metall Ital,1931,23:803-823.
[13]GSCHNEIDNER K A,CALDERWOOD F W.The Ag-La (Silver-Lanthanum) system[J].Bulletin of Alloy Phase Diagrams,1983,4(4):370-374.
[14]MCMASTERS O,GSCHNEIDER K,VENTEICHER R.Crystallography of the silver-rich rare-earth-silver intermetallic compounds[J].Acta Crystallographica Section B:Structural Crystallography and Crystal Chemistry,1970,26(9):1224-1229.
[15]STEEB S,GODEL D,L?HR C.On the structure of the compounds Ag 3 RE (RE=Y,La,Ce,Sm,Gd,Dy,Ho,Er)[J].Journal of the Less Common Metals,1968,15(2):137-141.
[16]FITZNER K,KLEPPA O.Thermochemistry of Binary Alloys of Transition Metals:The Systems Cu-Ce,Me-Pr,and Me-Nd (Me=Cu,Ag,Au)[J].Metallurgical and Materials Transactions A,1994,25(7):1495-1500.
[17]FITZNER K,JUNG W G,KLEPPA O J.Thermochemistry of binary alloys of transition metals:The Me-Sc,Me-Y,and Me-La (Me=Ag,Au) systems[J].Metallurgical & Materials Transactions A,1991,22(5):1103-1111.
[18]LI Z,SU X P,YIN F C,et al.On the thermodynamic assessment of the Ag-La system[J].Journal of Alloys and Compounds,2000,299(1/2):195-198.
[19]COBAN C,CIFTCI Y O,COLAKOGLU K.First principles investigations on the mechanical and vibrational properties for the selected B2-AgRE (RE=Sc,Y,La,Ce) intermetallics[J].Physica B:Condensed Matter,2015,457:22-29.
[20]SEGALL M,LINDAN P J,PROBERT M A,et al.First-principles simulation:ideas,illustrations and the CASTEP code[J].Journal of Physics:Condensed Matter,2002,14(11):2717.
[21]CLARK S J,SEGALL M D,PICKARD C J,et al.First principles methods using CASTEP[J].Zeitschrift für Kristallographie-Crystalline Materials,2005,220(5/6):567-570.
[22]VANDERBILT D.Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism[J].Physical Review B Condensed Matter,1990,41(11):7892-7895.
[23]FRANCIS G P,PAYNE M C.Finite basis set corrections to total energy pseudopotential calculations[J].Journal of Physics Condensed Matter,1998,2(19):4395-4404.
[24]PERDEW J P,BURKE K,ERNZERHOF M.Generalized Gradient Approximation Made Simple[J].Physical Review Letters,1997,78(7):1396.
[25]MONKHORST H J,PACK J D.Special points for Brillouin-zone integrations[J].Physical Review B,1976,13(12):5188-5192.
[26]FISCHER T H,ALMLOF J.General methods for geometry and wave function optimization[J].The Journal of Physical Chemistry,1992,96(24):9768-9774.
[27]SEKKAL A,BENZAIR A,OUAHRANI T,et al.Mechanical properties and bonding feature of the YAg,CeAg,HoCu,LaAg,LaZn,and LaMg rare-earth intermetallic compounds:An ab initio study[J].Intermetallics,2014,45:65-70.
[28]MAO P L,YU B,LIU Z,et al.First-principles calculations of structural,elastic and electronic properties of AB 2 type intermetallics in Mg-Zn-Ca-Cu alloy[J].Journal of Magnesium & Alloys,2013,1(3):256-262.
[29]RAY N,KEMPF B,MüTZEL T,et al.Effect of WC particle size and Ag volume fraction on electrical contact resistance and thermal conductivity of Ag-WC contact materials[J].Materials & Design,2015,85:412-422.
[30]KITTEL C.Introduction to solid state physics[M].Hoboken:John Wiley and Sons,2005.
(責任編輯楊文青)
Structural and Electronic Properties of Ag-La Binary Compounds from First-Principle Investigation
GAO En-qiang1, ZHANG Zhao-chao1, RUAN Hai-guang1,HUANG Fu-xiang1, CHEN Zhi-qian2, WANG Lan-lan1
(1.College of Material Science & Engineering, Chongqing University of Technology,Chongqing 400054, China; 2.Faculty of Materials and Energy,Southwest University, Chongqing 400715, China)
The first-principle calculation was performed to investigate the phase stability and electronic structure of four typical binary Ag-La compounds, including B2-LaAg, LaAg2, La14Ag51and α-LaAg5. The optimizedstructural parameters and mass densities of equilibrium were consistent with experimental data. The calculated cohesive energies indicate that the bond strength and stability of the four compounds is gradually enhanced with increasing La concentration. And enthalpies of formation of compounds are also consistent with experiment and CALPHAD calculation values, and their values are -21.7, -26.8, -22.9 and -18.1 kJ/mol, respectively. The results of electronic structure reveal that all Ag-La binary compounds analyzed here are conductors, and metallic, ionic and covalent bonds are involved. The ionic bond is attributed to charge moving from La atoms to Ag atoms, and the covalent bond is comprised of the hybridization of Ag s-p and Ag p-La d.With increasing La concentration in Ag-La compounds, both the strength of ionic and covalent bond are strengthened, leading the increasing of the stability of the compounds.
Ag-La system; rare earth phase; electronic structure; first-principle
2016-04-18
重慶市科技攻關項目(CSTC,2011AB4051)
高恩強(1988—),男,福建福清人,碩士研究生,主要從事功能材料及材料計算研究;通訊作者 黃福祥(1964—),男,重慶人,博士,教授,主要從事金屬功能材料、失效分析等研究,E-mail:hfuxiang@163.com。
format:GAO En-qiang, ZHANG Zhao-chao, RUAN Hai-guang, et al.Structural and Electronic Properties of Ag-La Binary Compounds from First-Principle Investigation[J].Journal of Chongqing University of Technology(Natural Science),2016(8):45-51.
10.3969/j.issn.1674-8425(z).2016.08.007
O641
A
1674-8425(2016)08-0045-07
引用格式:高恩強,張照超,阮海光,等.Ag-La系二元化合物結構與電子性能的第一性原理研究[J].重慶理工大學學報(自然科學),2016(8):45-51.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
