補正續骨丸質量標準研究
2016-09-22 05:40:19王作君馬宏達
世界中醫藥 2016年8期
賈 輝 顏 鳴 王作君 馬宏達
(沈陽軍區總醫院藥學部,沈陽,110016)
?
補正續骨丸質量標準研究
賈輝顏鳴王作君馬宏達
(沈陽軍區總醫院藥學部,沈陽,110016)
目的:建立并完善軍規制劑補正續骨丸的質量標準。方法:采用顯微鑒別法對處方中5味具有顯微鑒別特征的藥味進行鑒別;對方中續斷、何首烏、骨碎補、雞血藤、菟絲子運用薄層色譜法進行定性鑒別;樣品中柚皮苷的含量測定則運用專屬性較強的HPLC法進行定量測定。色譜條件:色譜柱為Welchrom C18柱(200 mm×4.6 mm,5 μm);流動相:乙腈-水(20∶80);流速為1.0 mL/min;檢測波長:283 nm;柱溫:25 ℃。結果:顯微鑒別特征明顯,薄層定性鑒別的斑點清晰,分離效果良好;柚皮苷在2.5~80 μg/mL濃度范圍內線性關系良好(r=1.0000),平均加樣回收率為96.06%,RSD為1.63%(n=6)。結論:本次研究所建立和完善的標準在方法上專屬性強,重現性好;在操作上簡便易行,省時省力。符合軍隊質量標準提升的要求,可作為該制劑列入新版軍規的質量標準。
補正續骨丸;質量標準;柚皮苷;HPLC;TLC
補正續骨丸由何首烏、續斷、骨碎補、雞血藤等11味中藥組成,具有補肝腎,活血脈,強筋壯骨,舒筋活絡,祛風除濕,消腫止痛之功效,主要用于治療骨質增生,頸椎綜合征,腰肌勞損、腰酸腿痛等。依據2013軍隊醫療機構制劑標準提高科研專項課題的總體要求,我們從校對制劑處方、明確制劑工藝各詳細參數、增加薄層鑒別藥味及建立健全含量測定項等各個方面對原軍規制劑補正續骨丸的質量標準進行提升,力求該制劑經此研究之后,質控水平與現行中國藥典同步。本課題在修訂原標準何首烏、菟絲子薄層鑒別的基礎上,增加了續斷、骨碎補和雞血藤的薄層鑒別;建立了方中骨碎補中指標性成分柚皮苷的含量測定。該軍規制劑標準經本次提高完善后,質量穩定,條件可控,更能保證患者安全用藥。
1 一般資料
高效液相(泵型號W600,紫外檢測器型號2487,Waters公司)。柚皮苷標準品(中國食品藥品檢定研究院,批號:110722-201312);續斷對照藥材(中國食品藥品檢定研究院,批號:121033-201311);大黃素對照品(中國食品藥品檢定研究院,批號:110756-200110);雞血藤對照藥材(中國食品藥品檢定研究院,批號:121173-201103);菟絲子對照藥材(中國食品藥品檢定研究院,批號:121232-201102);補正續骨丸(沈陽軍區司令部門診部,批號:20140220、20140625、20141120);水為重蒸水(本單位自制),乙腈(色譜級,Fisher公司),甲醇(色譜級,Fisher公司),其余試劑均為分析純。
2 方法與結果
2.1顯微鑒別1)雞血藤:草酸鈣簇晶,方晶較多,大小懸殊,稀有更大簇晶,呈不規則形,少數小方晶只10 μm左右,見圖1-1。導管多為具緣紋、網紋導管,見圖1-4。2)何首烏:單粒淀粉粒明顯可見,類球形粒,臍點呈星狀或三叉狀,見圖1-2。3)合歡:晶纖維粗大,見圖1-3。石細胞多成群、成無色或淡黃色、長方形或類圓形,見圖1-5。4)鹿茸:鏡下可見的顯微結構如:毛茸碎片,表面多為薄而透明的扁平細胞,見圖1-6。5)狗脊:鏡下可見的顯微結構如:金黃色的非腺毛,見圖1-7,1-8。
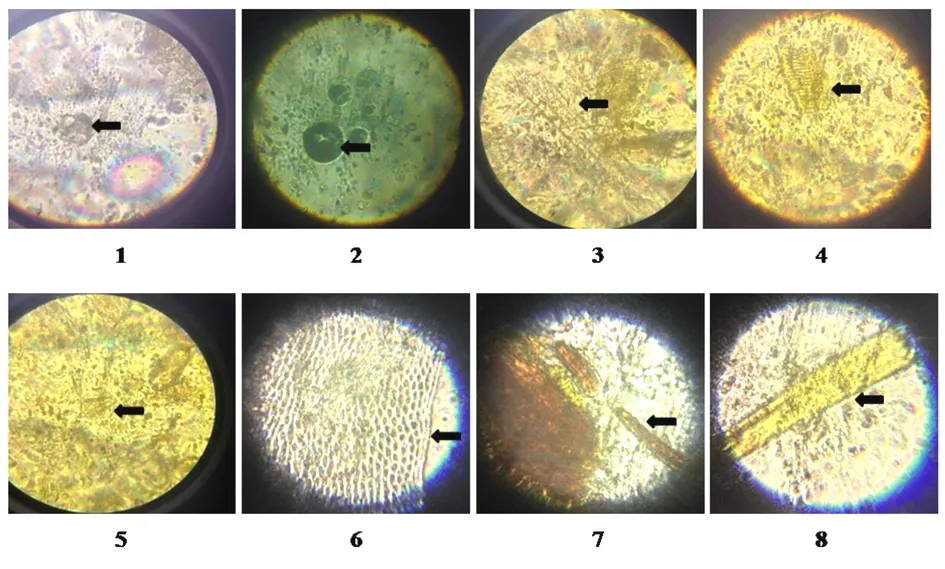
圖1 補正續骨丸顯微鑒別圖(10×100倍)
注:1.簇晶(雞血藤);2.淀粉粒(何首烏);3.晶纖維(合歡);4.導管(雞血藤);5.石細胞(合歡);6.毛茸碎片(鹿茸);7.金色非腺毛(狗脊);8.金色非腺毛(狗脊)。
2.2薄層鑒別
2.2.1續斷1)取補正續骨丸10 g,剪刀剪碎后研缽研細,加入40 mL分析甲醇,經30 min超聲提取,過濾,濾液水浴蒸干,殘渣使用蒸餾水30 mL溶解,水飽和正丁醇振搖提取,每次20 mL,重復3次,正丁醇液合并,用正丁醇飽和的1% NaOH堿液20 mL洗滌,重復2次,棄去堿液,再用正丁醇飽和的水20 mL洗滌1次,棄水液,正丁醇液通風櫥揮干,加乙醇1.0 mL溶解殘渣,即得供試品溶液[1]。取0.5 g續斷對照藥材,同法制成對照藥材溶液。取續斷陰性樣品蜜丸,按供試品溶液制備方法制得陰性樣品溶液。照薄層色譜法,分別吸取上述三種溶液各3 μL,點于一塊硅膠G薄層板,用(4∶1∶5)比例的正丁醇-醋酸-水的上層溶液[2]展開完畢后,通風櫥晾干,噴灑10%硫酸乙醇溶液,110 ℃加熱至斑點顯色清晰。供試品色譜與對照藥材斑點的色譜行為一致,陰性樣品色譜無干擾。見圖2A。
2.2.2何首烏取補正續骨丸15 g,剪刀剪碎后研缽研細,加入80 mL分析甲醇,經超聲處理40 min,過濾,濾液水浴蒸干,加蒸餾水20 mL溶解,用乙酸乙酯20 mL提取,重復3次,乙酸乙酯液合并,合并液水浴揮干,加甲醇1.0 mL使殘渣溶解,即得供試品溶液[3]。取大黃素對照品適量,加甲醇制成濃度1 mg/mL的對照品溶液。取何首烏陰性樣品蜜丸,按供試品溶液制備方法制得陰性樣品溶液。照薄層色譜法,分別吸取供試品和陰性樣品溶液各5 μL、對照品溶液2 μL,點于一塊硅膠G薄層板,用(20∶2∶1)比例的甲苯-乙酸乙酯-甲酸展開完畢后,通風櫥晾干,日光下檢視。供試品色譜與對照品斑點的色譜行為一致,顯同樣的黃色斑點,經氨氣熏蒸后斑點變為紅色,且陰性樣品色譜無干擾。見圖1B。
2.2.3骨碎補取補正續骨丸5 g,剪刀剪碎后研缽研細,加入20 mL分析甲醇,經30 min超聲處理,過濾,濾液水浴蒸干,殘渣加10 mL蒸餾水溶解,用20 mL乙酸乙酯提取1次,分離乙酸乙酯液水浴揮干,加甲醇1.0 mL使殘渣溶解,即得供試品溶液。取柚皮苷對照品,加甲醇制成濃度為0.5 mg/mL的對照品溶液。取骨碎補陰性樣品蜜丸,按供試品溶液制備方法制得陰性樣品溶液。照薄層色譜法,分別吸取上述三種溶液各10 μL,點于一塊硅膠G薄層板,用(1∶12∶2.5∶3)比例的甲苯-乙酸乙酯-甲酸-水的上層溶液[4]展開完畢后,通風櫥晾干,噴灑三氯化鋁試液顯色,置365 nm紫外光燈下檢視。供試品色譜與對照品斑點的色譜行為一致,且陰性樣品色譜無干擾。見圖2C。
2.2.4雞血藤取補正續骨丸10 g,剪刀剪碎后研缽研細,加入30 mL乙酸乙酯,經30 min超聲處理后過濾,濾液水浴揮干,加甲醇1.0 mL使殘渣溶解,即得供試品溶液。取0.5 g雞血藤對照藥材,同法制成對照藥材溶液。取雞血藤陰性樣品蜜丸,按供試品溶液制備方法制得陰性樣品溶液。照薄層色譜法,分別吸取上述三種溶液各5 μL,點于一塊硅膠G薄層板,用(30∶1)比例的二氯甲烷-甲醇展開完畢后,通風櫥晾干,置254 nm紫外光燈下檢視。供試品色譜與對照藥材斑點的色譜行為一致,且陰性樣品色譜無干擾。見圖2D。
2.2.5菟絲子取補正續骨丸10 g,剪刀剪碎后研缽研細,加入30 mL石油醚,經40 min超聲處理后,靜靜放置5 min后,傾倒出石油醚液,丟棄,殘渣置通風櫥揮干石油醚,加入60 mL甲醇,再次超聲處理40 min后濾過,濾液蒸干,殘渣加20 mL水使溶解,乙酸乙酯20 mL提取,重復2次,乙酸乙酯液合并,水浴揮干,殘渣加1.0 mL甲醇使溶解,即得供試品溶液[5]。取1 g菟絲子對照藥材,同法制成對照藥材溶液。取菟絲子陰性樣品蜜丸,按供試品溶液制備方法制得陰性樣品溶液。照薄層色譜法,分別吸取上述三種溶液各2 μL,點于一塊硅膠G薄層板,用(40∶30∶10∶7)比例的二氯甲烷-乙酸乙酯-甲醇-水的下層溶液展開完畢后,通風櫥晾干,噴灑三氯化鋁試液顯色,置365 nm紫外光燈下檢視。供試品色譜與對照藥材斑點的色譜行為一致,且陰性樣品色譜無干擾。見圖2E。
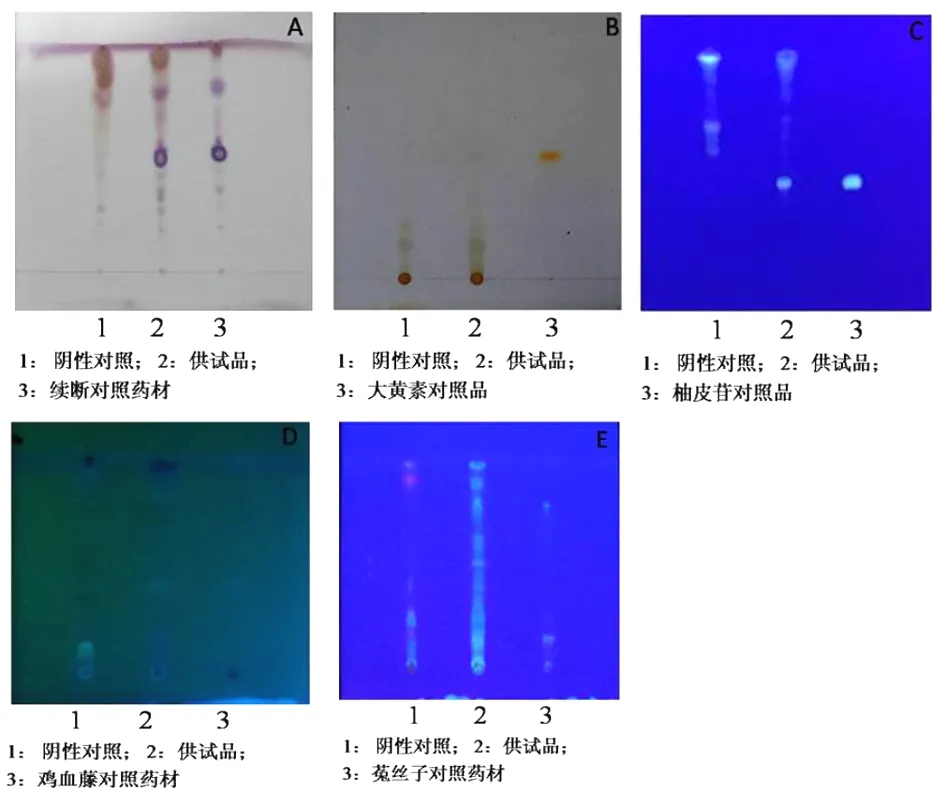
圖2 補正續骨丸TLC圖
注:A.續斷;B.何首烏;C.骨碎補;D.雞血藤;E.菟絲子。
2.3含量測定
2.3.1色譜條件色譜柱品牌為Welchrom C18柱,內徑與柱長為(200 mm×4.6 mm,5 μm);流動相組成為乙腈-水(20∶80);檢測波長設定為283 nm[6];流速設定為1 mL/min;柱溫設定:25 ℃;進樣量為20 μL。理論板數按柚皮苷峰計算高于3 000。
2.3.2溶液制備1)對照品溶液:柚皮苷對照品放置于干燥器中減壓干燥12 h以上后取適量,精密稱定,加入適量甲醇使溶解,制成濃度為30 μg/mL的柚皮苷對照品溶液。2)供試品溶液:取補正續骨丸2 g,剪刀剪碎后研缽研細,精密稱定,置圓底燒瓶中,精密加入40 mL 30%甲醇,稱定重量,水浴加熱回流提取1 h,放冷,再次稱定重量,用30%甲醇補足減失的重量,搖勻,濾過,離心(10 080 r/min)5 min,離心后取上清液2.5 mL置于10 mL容量瓶中,用30%甲醇定容至刻度,濾過,取續濾液,即得。3)陰性樣品溶液:取骨碎補陰性樣品蜜丸,按照供試品溶液制備方法制成陰性樣品溶液。
2.3.3專屬性試驗精密吸取制備好的供試品、對照品、陰性樣品3種溶液各20 μL,按色譜條件進樣測定。結果補正續骨丸供試品色譜與柚皮苷對照品色譜樣品峰色譜行為一致,且陰性樣品溶液不干擾主樣品峰的測定。柚皮苷峰保留時間約為15 min,分離度、對稱度、拖尾因子均符合要求,理論板數按柚皮苷峰計算高于3 000。試驗表明此法專屬性強。結果見圖3。
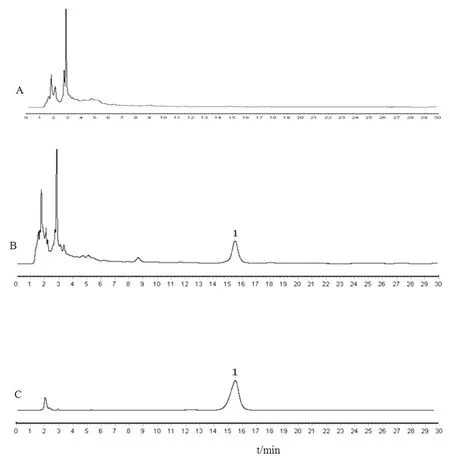
圖3 柚皮苷HPLC色譜圖
注:A.陰性溶液;B.供試品;C.對照品;1:柚皮苷。
2.3.4線性關系考察將柚皮苷對照品配制成濃度為2.5、5、10、20、40、80 μg/mL的溶液,精密吸取各溶液20 μL,按上述色譜條件進行測定,并繪制標準曲線。橫坐標為柚皮苷6個進樣濃度(μg/mL),縱坐標為各進樣濃度所測峰面積值,所得回歸方程:y=33 772x+7 853,r=1.000 0(n=6)。結果顯示,柚皮苷濃度在2.5~80 μg/mL范圍內與峰面積線性關系良好,所測補正續骨丸樣品落在線性范圍之中。
2.3.5儀器精密度考察柚皮苷對照品溶液,精密吸取20 μL,重復進樣6次,測定柚皮苷峰面積,計算RSD為0.15%。表明實驗所用HLPC精密度良好。
2.3.6樣品穩定性考察制備補正續骨丸供試品溶液一份(批號:20140220),室溫放置,在0、2、4、6、8、10、12 h分別進樣20 μL,測柚皮苷定峰面積。計算RSD為1.47%。表明補正續骨丸供試品溶液在12 h內穩定。
2.3.7方法重復性考察取補正續骨丸樣品(批號:20140220)6份,按供試品溶液制備方法制備并測定含量。計算得柚皮苷平均含量為1.204 2 mg/g,RSD為2.36%(n=6)。表明本方法重復性良好。
2.3.8回收率考察精密稱取已測知含量的補正續骨丸(批號:20140220)6份,每份約1 g,精密稱定,分別定量加入柚皮苷對照品,按供試品溶液方法制備,測定分析并計算回收率。結果見表1。
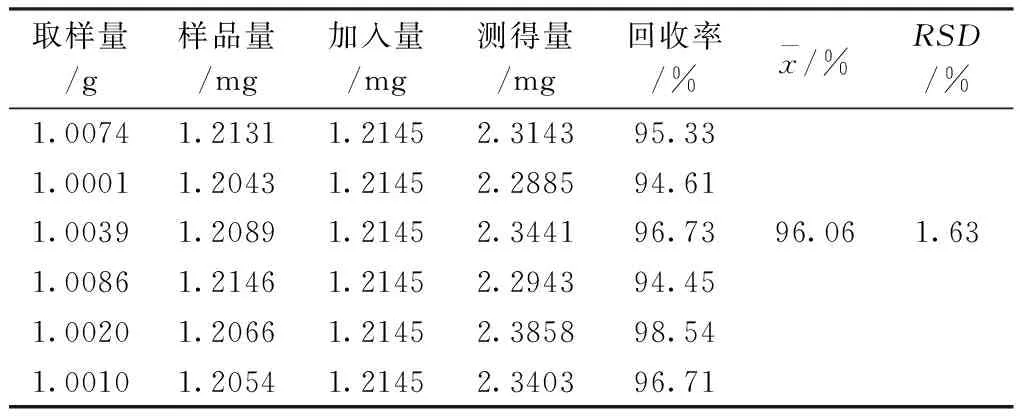
表1 柚皮苷加樣回收率試驗結果
2.3.9樣品含量測定取3批補正續骨丸樣品,精密稱定,按供試品溶液制備方法制備,進樣20 μL進行測定,用外標一點法計算補正續骨丸中柚皮苷含量。見表2。
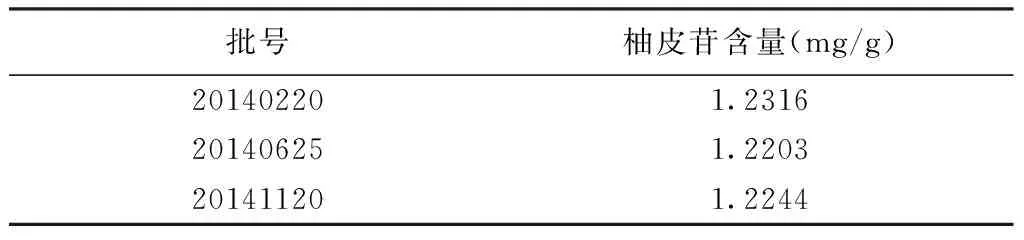
表2 樣品中柚皮苷含量測定結果(n=3)
2.3.10含量限度的確定根據對補正續骨丸三批中試樣品的含量測定,確定了該制劑的含量限度:本品每1 g含骨碎補以柚皮苷(C19H18O11)計,不得少于1.0 mg。
3 討論
本制劑處方有11味藥材,原標準中對何首烏、菟絲子2味藥材進行了薄層色譜鑒別,但二者重現性均較差。本研究在修訂何首烏、菟絲子薄層鑒別的基礎上,增加了續斷、骨碎補和雞血藤的薄層鑒別,經多次驗證,五味藥材的薄層鑒別方法專屬性及重復性均良好。
在整個實驗過程中,依據參考文獻的方法,本課題對供試品溶液制備的提取方式、提取溶劑、提取溶劑用量、提取時間等涉及到的諸多影響因素進行了考察和比較。對色譜條件中的流動相選擇也進行了不同比例的篩選,以求柚皮苷樣品峰的分離度、對稱度及拖尾因子均達到理想效果,理論板數合格,最終選擇了乙腈-水(20∶80)做為流動相,該流動相系統配制簡便,穩定性好,且陰性不干擾柚皮苷主峰的測定。
本試驗所建立的補正續骨丸質量標準從定性、定量2個方面共同驗證,力求工藝穩定,質量可靠,方法簡潔,操作順暢。最終通過對多批補正續骨丸樣品中柚皮苷含量的反復測定,分析計算,我們設定了該制劑的含量限度,以此作為該制劑今后實際檢驗工作的執行標準,也為該制劑藥效學研究的開展打下了堅實的基礎。
[1]許保海,臧琛,聶其霞.關節痛丸質量標準研究[J].中醫藥導報,2012,18(3):65-67.
[2]呂鵬,房德敏,趙薇,等.消腫散瘀膏質量標準研究[J].天津藥學,2009,21(4):18-19.
[3]劉繼華,李曼杰,王威,等.薄層色譜法鑒別蟻王壯力膠囊中的黑螞蟻菟絲子何首烏[J].時珍國醫國藥,1998,9(5):431.
[4]陳英紅,黃恩喜,高陽,等.抗骨增生丸定性定量方法研究[J].中國實驗方劑學雜志,2006,12(7):20-23.
[5]熊建國,左斌.金剛丸質量標準考察[J].中成藥,2008,30(7):附15-附17.
[6]商麗華,龍海東,方樂堯,等.骨仙片質量標準提高研究[J].中成藥,2010,32(2):335-337.
(2015-08-26收稿責任編輯:張文婷)
Quality Standard Study on Buzheng Xugu Pill
Jia Hui,Yan Ming,Wang Zuojun,Ma Hongda
(DepartmentofPharmacyofGeneralHospitalofShenyangMilitaryCommandArea,Shenyang110016,China)
Objective:To establish a quality standard for Buzheng Xugu pill.Methods:Five crude drugs in the prescription were identified by microscope,including Duanxu (Radix Dipsaci),Heshouwu (Polygonum multiflorum),Gusuibu (Rhizoma Drynariae),Jixueteng (Caulis Spatholobi) and Tusizi (Semen Cuscutae) were identified by TLC method.Also,the content of Naringin in Buzheng Xugu pill was determined by HPLC.The Welchrom C18(200 mm×4.6 mm,5 μm) column was used.The mobile phase consisted of acetonitrile-water (20:80) was at a flow rate of 1.0 mL/min,and the detection wavelength was at 283 nm under 25 ℃.Results:The microscopic features were easy to identify:the spots in the thin layer chromatography were clear.The Naringin showed a good linear relationship within the range of 2.5~80 μg/mL ( r=1.0000).The average recovery was 96.06%,and theRSDwas 1.63%(n=6).Conclusion:This method is simple,reliable,reproducible and can be used in the quality control of the preparation of the drug.
Buzheng Xugu pill; Quality standard; Naringin; HPLC; TLC
軍隊醫療機構制劑標準提高科研專項課題——遼沈地區預防治療常見軍事訓練傷醫院制劑的標準提升研究(編號:14ZJZ13-1)
賈輝(1982.12—),中國人民解放軍沈陽軍區總醫院藥學部,藥師,碩士,研究方向:中藥學,Tel:(024)28851751,E-mail:smockingandy@163.com
馬宏達(1976.09—),男,博士,主管藥師,研究方向:藥劑學,Tel:(024)28851750,E-mail:mahongdamahongda@163.com
R284.1
A
10.3969/j.issn.1673-7202.2016.08.054
