伴剝脫性皮炎的噬血細胞綜合征一例
2016-11-06 03:05:47王丹吳景良李雪飛陸海濤段昕所
中華皮膚科雜志 2016年9期
王丹 吳景良 李雪飛 陸海濤 段昕所
067000 河北,承德醫(yī)學院附屬醫(yī)院皮膚科
伴剝脫性皮炎的噬血細胞綜合征一例
王丹 吳景良 李雪飛 陸海濤 段昕所
067000 河北,承德醫(yī)學院附屬醫(yī)院皮膚科
患者女,75歲。因全身彌漫性潮紅、脫屑伴瘙癢2個月就診。2個月前患者無明顯誘因全身出現彌漫性潮紅、脫屑,逐漸加重,自覺皮疹處疼痛。10余天前患者無誘因出現白細胞降低,1 d前患者出現發(fā)熱,體溫高達39℃,前胸及顏面部嚴重紅腫伴喘息、氣短,以剝脫性皮炎收入院。發(fā)病以來,患者精神一般,睡眠差,食欲可,大小便正常,體重下降7.5 kg,體力下降。既往史:患有慢性支氣管炎病史20余年,曾間斷口服中藥(具體不詳)治療。個人史及家族史無特殊。
體檢:體溫36.3℃,脈搏96次,呼吸20次,血壓104/60 mmHg(1 mmHg=0.133 kPa),慢性病容,表情痛苦,右側頜下可觸及腫大淋巴結,雙肺呼吸音粗,可聞及少許干濕性啰音。雙足輕度凹陷性水腫。皮膚科檢查:全身彌漫性潮紅、浮腫,紅斑表面干燥,可見大量細小片狀脫屑,見圖1。黏膜無損害,毛發(fā)、指(趾)甲未見異常。
實驗室檢查:入院后監(jiān)測血常規(guī)變化。白細胞計數最低0.1×109/L,血小板計數最低16×109/L。血生化檢查:總膽紅素 36.5 μmol/L(參考值 3~22 μmol/L),白蛋白 26 g/L(35~50 g/L),鉀2.53 mmol/L(3.5~5.5 mmol/L),鈣1.85 mmol/L(2~2.7 mmol/L),乳酸脫氫酶805 U/L(300~620 U/L)。降鈣素原0.34 μg/L(< 0.5 μg/L)。胸部CT:兩肺陳舊性病變,右肺上葉膨脹不全,右肺中葉不張。右側胸腔積液,左肺葉間積液。D-二聚體升高至6.21~9.7 mg/L(0~0.55 mg/L),纖維蛋白原下降至0.91~1.479 mg/L(0~5 mg/L)。血清鐵蛋白>2 000 μg/L(13~ 150 μg/L)。骨髓鐵染色:外鐵++(+ ~++),內鐵48%陽性(19%~44%陽性)。染色體分析未見分裂象。骨髓象:增生活躍,粒系增生降低,組織細胞增多占0.025,可見吞噬血細胞現象(圖2)。取背部紅斑脫屑處做皮膚病理:角化不全,真皮乳頭水腫,真皮淺層血管周圍散在單個核細胞浸潤。
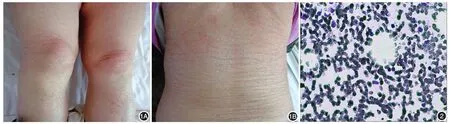
圖1 患者皮膚彌漫潮紅,浮腫,表面見細小片狀鱗屑 1A:雙下肢;1B:背部 圖2 骨髓象 組織細胞增多,可見吞噬血細胞現象
診斷及治療:①噬血細胞綜合征(HPS);②剝脫性皮炎;③彌散性血管內凝血(DIC);④炎癥急性加重期;⑤低鉀血癥、低蛋白血癥、血小板減少癥、粒細胞減少癥;⑥右肺中葉不張、右側胸腔積液。治療:給予抗炎抗過敏,補鉀,補液,補充白蛋白,外用保濕劑尿囊素治療。患者間斷出現發(fā)熱、喘息、呼吸困難,給予舒巴坦+頭孢哌酮、美羅培南抗感染,甲潑尼龍琥珀酸鈉抗炎,多索茶堿舒張氣道,粒細胞集落刺激因子升白細胞,輸同型冷沉淀凝血因子改善凝血,輸冰凍單采少白細胞血小板升血小板等治療。經上述治療,患者呼吸系統(tǒng)癥狀、皮疹曾一度好轉,但血小板、白細胞計數、纖維蛋白原仍持續(xù)下降。患者喘息、呼吸困難逐漸加重,精神狀況差,四肢濕冷,口唇發(fā)紺,雙肺呼吸音粗,可聞及干濕性啰音,全身皮膚可見多發(fā)瘀點瘀斑。期間多次請呼吸科、血液科等多科室會診,后轉入血液科治療,轉科后患者呼吸困難進行性加重、出現昏迷,死亡。
討論HPS是一組由多種原因引起的從骨髓細胞角度提出的綜合征,伴有組織細胞活躍吞噬自身各種血細胞的現象[1]。分為原發(fā)性和繼發(fā)性,其中原發(fā)性以嬰幼兒多見,是常染色體隱形遺傳性疾病[2],而繼發(fā)性可見于各年齡階段,但以成人更多見[3]。HPS臨床上多表現為高熱,肝、脾淋巴結腫大,全血細胞減少和凝血障礙,病死率極高,抗生素及糖皮質激素治療效果不佳。
檢索CHKD期刊全文數據庫(1995—2016年),剝脫性皮炎繼發(fā)于HPS尚未見報道。本例患者以剝脫性皮炎為首發(fā)表現,可能發(fā)生于HPS后,也可能是HPS的繼發(fā)表現,繼之HPS病情迅速進展終導致死亡,結合實驗室檢查、臨床癥狀及既往用藥史,其剝脫性皮炎和HPS亦不排除由腫瘤或藥物所誘發(fā)。但患者病情進展極快,一般情況差,隨時可自發(fā)大出血、DIC、循環(huán)衰竭而危及生命,行支氣管鏡、胸腔穿刺、腹腔穿刺風險極大,因此未能找到確切腫瘤證據。HPS十分兇險,診斷困難,患者預后極差,只有對其有足夠充分的認識,才能做到早期診斷及治療,這對提高患者的生存率十分重要。
[1]Henter JI,Horne A,Aricó M,et al.HLH?2004:diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis[J].Pediatr Blood Cancer,2007,48(2):124?131.DOI:10.1002/pbc.21039.
[2]Gholam C,Grigoriadou S,Gilmour KC,et al.Familial haemophago?cytic lymphohistiocytosis:advances in the genetic basis,diagnosis andmanagement[J].Clin Exp Immunol,2011,163(3):271 ?283.DOI:10.1111/j.1365?2249.2010.04302.x.
[3]Janka GE.Familial and acquired hemophagocytic lymphohistio?cytosis[J].Annu Rev Med,2012,63:233 ?246.DOI:10.1146/annurev?med?041610?134208.
2015?09?21)
(本文編輯:尚淑賢)
段昕所,Email:duanxinsuo2002@163.com
10.3760/cma.j.issn.0412?4030.2016.09.021
