丹磺酰氯柱前衍生-超高效液相色譜-串聯(lián)質(zhì)譜法測(cè)定人體尿樣中的環(huán)己胺
2016-11-09 08:32:51楊華梅杭莉楊建國(guó)張雪梅黃為紅李晨蔣曉麗
分析化學(xué) 2016年3期
楊華梅杭莉 楊建國(guó) 張雪梅 黃為紅 李晨 蔣曉麗
(泰州市疾病預(yù)防控制中心,泰州225300)
評(píng)述與進(jìn)展
丹磺酰氯柱前衍生-超高效液相色譜-串聯(lián)質(zhì)譜法測(cè)定人體尿樣中的環(huán)己胺
楊華梅"杭莉楊建國(guó)張雪梅黃為紅李晨蔣曉麗
(泰州市疾病預(yù)防控制中心,泰州225300)
建立丹磺酰氯柱前衍生-超高效液相色譜-串聯(lián)質(zhì)譜法測(cè)定人體尿樣中環(huán)己胺的方法。冷凍樣品經(jīng)解凍、離心后,用丹磺酰氯衍生,固相萃取小柱凈化。目標(biāo)化合物采用Waters ACQUITY CSHTMC18色譜柱(50 mm×2.1 mm,1.7μm)分離,以甲醇和0.002mol/L乙酸銨溶液為流動(dòng)相梯度洗脫,采用電噴霧離子源電離、正離子多反應(yīng)監(jiān)測(cè)模式質(zhì)譜檢測(cè)。環(huán)己胺在2.5~200μg/L濃度范圍內(nèi)有較好的線性關(guān)系,相關(guān)系數(shù)大于0.999,回收率為98.7%~102.3%,精密度為3.1%~5.2%,檢出限和定量限分別為1.0和3.0μg/L。結(jié)果表明,本方法操作簡(jiǎn)單、準(zhǔn)確可靠,可適用于人體尿液中環(huán)己胺的定量分析。應(yīng)用本方法測(cè)定200份學(xué)生尿液樣品,環(huán)己胺檢出率為34.5%。
環(huán)己胺;丹磺酰氯;衍生;固相萃取;人體尿樣;超高效液相色譜-串聯(lián)質(zhì)譜
1 引言
甜蜜素是第三代無(wú)熱量的代糖食品添加劑[1],化學(xué)名稱為環(huán)己基氨基磺酸鈉,屬于合成甜味劑,分子式為C6H11NH-SO3Na,英文名為Sodium Cyclamate,是食品生產(chǎn)中常用的添加劑,其口感好、價(jià)格低廉,甜度是蔗糖的30~40倍[2]。自1970年P(guān)rice等[3]報(bào)道甜蜜素對(duì)大鼠可誘發(fā)膀胱癌后,研究者對(duì)甜蜜素的毒理及甜蜜素體內(nèi)代謝機(jī)理展開(kāi)了長(zhǎng)期研究[4~9]。研究表明,甜蜜素在人體內(nèi)不能被新陳代謝,由尿液和糞便排出,但有些人腸內(nèi)細(xì)菌可使之降解為比甜蜜素毒性大的致癌物質(zhì)環(huán)己胺(Cyclohexylamine)[10]。目前,世界各國(guó)對(duì)甜蜜素的安全性仍存在爭(zhēng)議,至今中國(guó)、德國(guó)等80多個(gè)國(guó)家明確準(zhǔn)用,而日本、美國(guó)、英國(guó)等45個(gè)國(guó)家明確禁用[11]。
盡管我國(guó)對(duì)甜蜜素有嚴(yán)格的限量標(biāo)準(zhǔn),但許多生產(chǎn)廠家卻未嚴(yán)格執(zhí)行國(guó)家標(biāo)準(zhǔn),加上相關(guān)的執(zhí)法力度不夠,導(dǎo)致超標(biāo)濫用的情況屢屢出現(xiàn)[11,12]。甜蜜素在我們飲食生活中無(wú)處不在,尤其零食中居多,而兒童最喜愛(ài)吃甜味零食,經(jīng)常食用含有甜蜜素超標(biāo)的飲料或其它食品,就會(huì)因其攝入過(guò)量而對(duì)代謝排毒能力較弱的兒童身體造成危害,因此,對(duì)兒童尿液中的環(huán)己胺含量測(cè)定尤為重要。1971年P(guān)rosky等[13]為了解甜蜜素代謝成環(huán)己胺的比例狀況,采用紙電泳方法測(cè)定幼鼠尿液中甜蜜素和環(huán)己胺的含量; 1972年Renwick等[14]為了解環(huán)己胺在體內(nèi)的代謝機(jī)理,采用紙色譜方法測(cè)定人體、老鼠、兔子和豬等尿液中的環(huán)己胺及其相應(yīng)的代謝物含量;1976年Matsumura等[15]采用氣相色譜法同時(shí)測(cè)定尿液中環(huán)己胺和N-甲基環(huán)己胺;Isidre[16]和Renwick[9]等分別于1996年和2004年采用2,4,6-三硝基苯磺酸柱前衍生液相色譜法對(duì)人體尿液中甜蜜素和環(huán)己胺的含量進(jìn)行了測(cè)定,而我國(guó)尚未開(kāi)展對(duì)人體尿液中環(huán)己胺的檢測(cè)方法和含量調(diào)查。本研究建立了人體尿液中環(huán)己胺的超高效液相色譜-串聯(lián)質(zhì)譜法,與文獻(xiàn)報(bào)道的柱前衍生液相色譜法相比,本方法前處理采用了固相萃取小柱去除過(guò)量的衍生劑,且質(zhì)譜定性分析避免了假陽(yáng)性結(jié)果的出現(xiàn),此方法簡(jiǎn)單易操作,靈敏度高,達(dá)到了定性和定量分析的要求。
2 實(shí)驗(yàn)部分
2.1儀器與試劑
Waters ACQUITY超高效液相色譜;AB SCIEX API4000+質(zhì)譜儀;中佳HC-3018R高速冷凍離心機(jī);Poly-Sery MCX固相萃取柱(60 mg,3 mL,美國(guó)Supelco公司)。甲醇和丙酮(質(zhì)譜級(jí),德國(guó)Merck公司);實(shí)驗(yàn)用水:屈臣氏瓶裝蒸餾水;乙酸銨(色譜純,美國(guó)Tedia公司);HCl,NH3·H2O,NaOH和NaHCO3(分析純,天津博迪化工有限公司);丹磺酰氯(上海Sigma-Aldrich公司)。環(huán)己胺標(biāo)準(zhǔn)品(純度>99%,美國(guó)Supelco公司);D11-環(huán)己胺(25 mg,Toronto Research Chemicals公司)。丹磺酰氯衍生劑(2 mg/mL):準(zhǔn)確稱取0.1000 g丹磺酰氯,用丙酮溶解并定容至50 mL,現(xiàn)用現(xiàn)配。NaHCO3緩沖液:稱取4.2 g NaHCO3,溶于1000 mL水中,用0.1 mol/L NaOH調(diào)至pH 11.5。
2.2實(shí)驗(yàn)條件
2.2.1色譜條件Waters ACQUITY CSHTMC18色譜柱(50 mm×2.1 mm,1.7μm);流速:0.2 mL/min;進(jìn)樣體積:10.0μL;柱溫:40℃;樣品室溫度:25℃;流動(dòng)相:A為0.002 mol/L乙酸銨溶液;B為甲醇;梯度洗脫程序:0~4 min,10%~90%B;4~5.5 min,90%B;5.5~6 min,90%~10%B;6~8 min,90%B。
2.2.2質(zhì)譜條件掃描方式:電噴霧電離源(ESI+)掃描;采集模式:多反應(yīng)監(jiān)測(cè)(MRM)模式;碰撞氣壓力(CAD):4 Pa;氣簾氣壓力(CUR):30 Pa;霧化氣壓力(GS1):55 Pa;輔助氣壓力(GS2): 60 Pa;入口電壓(EP):10 V;碰撞室出口電壓(CXP):12 V;噴霧電壓:5500 V;氣化溫度:500℃。定量和定性離子,碰撞能量等參數(shù)見(jiàn)表1。
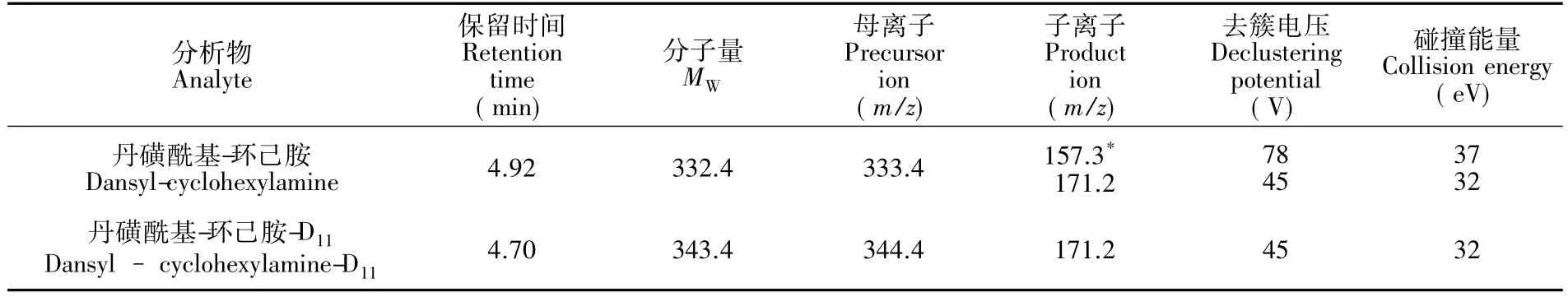
表1 保留時(shí)間,相對(duì)分子質(zhì)量和優(yōu)化的質(zhì)譜分析參數(shù)Table 1 Retention time,relativemolecular weight(MW)and optimized MS/MS parameters
2.3標(biāo)準(zhǔn)溶液的配制
標(biāo)準(zhǔn)品儲(chǔ)備液:分別準(zhǔn)確稱取環(huán)己胺和D11-環(huán)己胺各0.1 g(精確至0.1 mg),分別用水溶解并稀釋至100 mL,配成1.0 mg/mL標(biāo)準(zhǔn)儲(chǔ)備液,于4℃保存。標(biāo)準(zhǔn)工作液:分別準(zhǔn)確移取適量環(huán)己胺和D11-環(huán)己胺準(zhǔn)品儲(chǔ)備液于容量瓶中,加水稀釋,配成1.0μg/mL標(biāo)準(zhǔn)工作液。
2.4樣品制備
尿液樣品采集后于-20℃冷凍保存,臨用于室溫下緩慢解凍,以5000 r/min離心5 min,取上清液衍生后測(cè)定。
2.5樣品衍生和凈化
取2.4節(jié)上清液100μL,加水900μL,D11-環(huán)己胺標(biāo)準(zhǔn)工作液20μL,丹黃酰氯溶液0.5mL,NaHCO3緩沖液1.0 mL,充分混勻,于60℃水浴加熱15 min,取出冷卻至室溫,加入2 mL水稀釋,用0.5 mol/L HCl調(diào)至pH≈3。將樣液過(guò)活化后的Poly-Sery MCX固相萃取小柱,去除初濾液,用6 mL水沖洗抽干,用1 mL甲醇-氨水(95∶5,V/V)溶液洗脫,洗脫液經(jīng)氮?dú)獯蹈桑詈笥? mL甲醇溶解后,過(guò)0.2μm濾膜,待測(cè)。取 2.5,5.0,10,20,50,100和 200μL環(huán)己胺標(biāo)準(zhǔn)工作液于具塞試管中,各加100μL空白尿樣和適量水至1 mL后,按樣品處理方法處理。
3 結(jié)果與討論
3.1衍生化條件的考察
在探究衍生試劑時(shí)發(fā)現(xiàn),用丙酮或乙腈作為溶劑時(shí),衍生劑濃度高,會(huì)出現(xiàn)飽和不溶等現(xiàn)象,因此丹磺酰氯濃度確定為2 mg/mL。在優(yōu)化衍生條件的實(shí)驗(yàn)中,全部采用0.2μg/mL環(huán)己胺為分析物濃度。
3.1.1衍生劑體積丹磺酰氯和環(huán)己胺反應(yīng)生成磺酰胺類化合物,該反應(yīng)不能進(jìn)行完全,因此通常采用過(guò)量的丹磺酰氯對(duì)環(huán)己胺進(jìn)行衍生,盡可能將環(huán)己胺定量轉(zhuǎn)化成相應(yīng)的磺酰胺化合物。本實(shí)驗(yàn)考察了丹磺酰氯體積分別為0.05,0.10,0.25,0.50,1.00和2.00 mL時(shí)的環(huán)己胺衍生效率。實(shí)驗(yàn)表明,在pH=11.5,60℃反應(yīng)15 min的條件下,隨著丹磺酰氯體積增大,衍生物的轉(zhuǎn)化率也逐漸增大,當(dāng)衍生劑體積≥0.5 mL時(shí),轉(zhuǎn)化率基本保持不變,故衍生試劑體積選定為0.5 mL。
3.1.2pH值丹磺酰氯與環(huán)己胺反應(yīng)生成磺酰胺類化合物和氯化氫,反應(yīng)體系為堿性時(shí),可以提高反應(yīng)速率,實(shí)驗(yàn)中比較了緩沖液pH值(9.0,10.0,11.0和11.5)對(duì)衍生效率的影響,結(jié)果表明,緩沖液pH值越大,衍生效率越高,故選擇緩沖溶液pH=11.5。
3.1.3衍生溫度分別在30℃,40℃,60℃,70℃和90℃的條件下考察溫度對(duì)衍生反應(yīng)轉(zhuǎn)化效率的影響,實(shí)驗(yàn)表明,30℃時(shí)衍生效率最低,在40℃~70℃之間溫度對(duì)衍生效率無(wú)明顯差別,而在90℃時(shí),反應(yīng)溫度過(guò)高,導(dǎo)致溶劑揮發(fā),從而使衍生效率下降,故選擇溫度為60℃。
3.1.4衍生時(shí)間在60 min內(nèi)研究了反應(yīng)時(shí)間對(duì)衍生效率的影響。結(jié)果表明,隨著反應(yīng)時(shí)間的延長(zhǎng),轉(zhuǎn)化效率逐漸增大,在反應(yīng)15 min后,衍生反應(yīng)達(dá)到平衡,衍生產(chǎn)物的濃度基本保持不變。故衍生時(shí)間選擇為15 min。
與文獻(xiàn)中報(bào)道的2,4,6-三硝基苯磺酸衍生方法[16]相比,此衍生方法簡(jiǎn)單方便、時(shí)間短,能夠滿足尿液中環(huán)己胺的分析測(cè)定。
3.2固相萃取條件的選擇
比較了CNWBOND LC-C18固相萃取柱(500mg,6mL)和Poly-Sery MCX固相萃取柱(60mg,3 mL),結(jié)果表明,通用的C18萃取小柱不能將待測(cè)物和衍生劑分離,導(dǎo)致大量衍生劑水解產(chǎn)物一并洗脫,進(jìn)而污染色譜柱和離子源;而MCX萃取小柱為混合型陽(yáng)離子柱,適合吸附堿性化合物,能夠去除大量衍生劑。因此,本實(shí)驗(yàn)選擇了專用型的Poly-Sery MCX固相萃取柱(60 mg,3 mL)。
3.3衍生物的穩(wěn)定性
配制5和50 ng/mL的環(huán)己胺標(biāo)準(zhǔn)溶液,衍生后得到的衍生產(chǎn)物分別于0,2,4,8,10,24和48 h測(cè)定,結(jié)果表明,環(huán)己胺的丹黃酰氯衍生物在室溫下放置48 h內(nèi)穩(wěn)定。
3.4液相色譜條件的優(yōu)化
本研究采用Waters ACQUITY CSHTMC18色譜柱,以甲醇和乙酸銨溶液為流動(dòng)相,在水中加入0.002 mol/L乙酸銨有助于待測(cè)物良好峰型的形成,并能增強(qiáng)檢測(cè)的靈敏度。對(duì)基質(zhì)標(biāo)準(zhǔn)溶液衍生后進(jìn)行分析,甲醇相起始比例為10%,最終比例為90%,運(yùn)行8 min,梯度洗脫,能夠滿足待測(cè)物測(cè)定的需要。環(huán)己胺衍生物的總離子流圖如圖1所示。
3.5質(zhì)譜條件的優(yōu)化
3.5.1定性離子與定量離子的選擇根據(jù)待測(cè)物的結(jié)構(gòu)特征,選擇正離子模式電離,采用流動(dòng)注射泵進(jìn)樣,分別對(duì)1.0 mg/L的環(huán)己胺及D11-環(huán)己胺的丹磺酰氯衍生物進(jìn)行一級(jí)質(zhì)譜掃描,確定其準(zhǔn)分子離子峰,并分別優(yōu)化其去簇電壓。分別以其準(zhǔn)分子離子為母離子,通過(guò)碰撞氣碰撞產(chǎn)生碎片離子進(jìn)行二級(jí)質(zhì)譜掃描,同時(shí)優(yōu)化碰撞電壓,選擇豐度較高的2種碎片離子作為定性與定量特征離子。通過(guò)實(shí)驗(yàn)優(yōu)化,待測(cè)物的定性與定量特征離子及優(yōu)化的質(zhì)譜參數(shù)見(jiàn)表1。
3.5.2反應(yīng)方程式和質(zhì)譜裂解途徑丹黃酰氯和環(huán)己胺在堿性條件下加熱脫去一個(gè)HCl分子,生成環(huán)己丹磺酰胺。圖2中碎裂途徑表明,環(huán)己丹磺酰胺在流動(dòng)相乙酸銨的作用下形成m/z 333.4母離子,環(huán)己丹磺酰胺正離子有兩種裂解方式,一是CS單鍵的斷裂脫去環(huán)己胺磺基形成m/z 171.2子離子,二是脫去環(huán)己胺磺基和一個(gè)亞甲基形成m/z 157.3子離子。
3.6方法學(xué)考察
向空白尿樣中加入系列混合標(biāo)準(zhǔn)溶液,衍生凈化后在優(yōu)化的實(shí)驗(yàn)條件下測(cè)定。根據(jù)標(biāo)準(zhǔn)工作曲線測(cè)定結(jié)果,定量離子的峰面積比(Y)為縱坐標(biāo),質(zhì)量濃度為橫坐標(biāo)X(μg/L),進(jìn)行線性回歸線分析,在2.5~200μg/L范圍內(nèi),待測(cè)物峰面積與濃度量較好的線性關(guān)系,Y=2.2080×106X+16776,待測(cè)物的相關(guān)系數(shù)大于0.999。化合物定性檢出限(LOD)為樣品陰性基質(zhì)中添加標(biāo)準(zhǔn)品的方式獲得(S/N≥3),定量檢出限(LOQ)為樣品陰性基質(zhì)中添加標(biāo)準(zhǔn)品的方式獲得(S/N≥10),分別為1和3μg/L。與文獻(xiàn)[15,16]方法相比,本方法具有更低的檢出限(表2)。
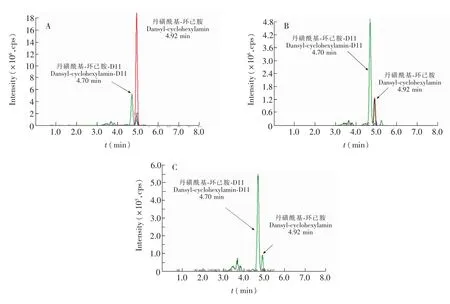
圖1 環(huán)己胺(50μg/L)及D11-環(huán)己胺(20μg/L)標(biāo)準(zhǔn)(A)、陽(yáng)性樣品(B)和空白尿樣(C)丹磺酰氯衍生物的總離子流圖Fig.1 Total ion current chromatogram of dansyl chloride derivatives of cyclohexylamine(50μg/L)and cyclohexylamine-D11(20μg/L)(A),positive sample(B)and blank urine(C)

圖2 待測(cè)物的碎裂途徑Fig.2 Fragmentation pathways for analyte
為了考察方法的重現(xiàn)性,添加2個(gè)濃度水平的環(huán)己胺到空白尿樣中進(jìn)行衍生凈化,對(duì)1 d內(nèi)獨(dú)立處理的樣品進(jìn)行6次測(cè)定,計(jì)算日內(nèi)回收率(RSD);以連續(xù)6 d獨(dú)立處理的樣品進(jìn)行測(cè)定,計(jì)算日間回收率(RSD);結(jié)果如表3所示。結(jié)果表明,本方法具有較好的重現(xiàn)性。

表2 本方法與其它文獻(xiàn)方法檢出限的比較Table 2 Comparison of LOD among differentmethods for determination of cyclohexylamine

表3 空白尿樣中不同環(huán)己胺加標(biāo)濃度的回收率和精密度Table 3 Recoveries and precisions of two different concentrations cyclohexylamine added to blank urine(n=6)
3.7實(shí)際樣品分析
應(yīng)用本方法分析200份當(dāng)?shù)匦W(xué)生尿樣,在所有檢測(cè)的樣品中環(huán)己胺的總檢出率為34.5%,檢出濃度在0.01~37.5 mg/L之間;其中131份尿樣環(huán)己胺濃度低于檢出限,35份尿樣檢測(cè)濃度在0.01~0.09 mg/L,21份尿樣檢測(cè)濃度在0.11~0.79 mg/L,13份尿樣檢測(cè)濃度在1.2~37.5 mg/L。與報(bào)道的文獻(xiàn)相比,本方法靈敏度高,環(huán)己胺的檢出率也相應(yīng)提高。檢測(cè)結(jié)果顯示,甜蜜素在人體是否代謝成環(huán)己胺以及環(huán)己胺的代謝濃度也可能是因人而異的,這與報(bào)道的文獻(xiàn)相符。
4 結(jié)論
本研究將丹磺酰氯柱前衍生-液相色譜-質(zhì)譜方法應(yīng)用于人體尿液中甜蜜素的代謝物環(huán)己胺的測(cè)定,衍生過(guò)程簡(jiǎn)單快速,并且混合型陽(yáng)離子固相萃取小柱較好地去除了過(guò)量的衍生劑和尿液基質(zhì)干擾,結(jié)合高效液相色譜質(zhì)譜檢測(cè),成功分析了尿樣中環(huán)己胺的含量。結(jié)果表明,本方法具有檢出限低、重現(xiàn)性好、回收率高、操作簡(jiǎn)便等特點(diǎn),可為甜蜜素的代謝毒理研究和評(píng)估人體環(huán)己胺暴露量提供一種簡(jiǎn)單靈敏、穩(wěn)定可靠的分析方法。
References
1YU Guo-Xin.China Food Safety,2011,10:48-49余國(guó)新.食品安全導(dǎo)刊,2011,10:48-49
2HAN Hua,JI Yuan-Bing,LI Jie-Li,YOU Ji-Ming,LI Hong-Mei,YANG Kang-Zhen.Green Science and Technology,2013,3:148-150韓華,紀(jì)元兵,李潔莉,尤繼明,李紅梅,楊抗震.綠色科技,2013,(3):48-150
3Price JM,Biava CG,Oser B L,Vogin E E,Steinfeld J,Ley H L.Science.,1970,167:1131-1132
4Mohd I,Deepak M,Shaik N R,Sudhir K S,Tulsidas R B.J.Pharm.Bioallied.Sci.,2013,5(1):44-48
5Buss N E,Renwick A G,Donaldson K M,George C F.Toxicol.Appl.Pharmacol.,1992,115:199-210
6Takayama S,Renwick A G,Johansson S L,Thorgeirsson U P,Tsutsumi M,Dalgard D W,Sieber SM.Toxicol.Sci.,2000,53:33-39
7Serra-Majem L,Bassas L,García-Glosas R,Ribas L,Inglés C,Casals I,Saavedra P,Renwick AG.Food AdditContam.,2003,20:1097-1104
8Andreatta M M,Muoz SE,LantieriM J,Eynard A R,Navarro A.Prev.Med.,2008,47:136-139
9Renwick A G,Thompson JP,O'Shaughnessy M,Walteret E J.Toxicol.Appl.Pharmacol.,2004,196:367-380
10Drassr B S,Renwick A G,Williams R T.J.Biochem.,1972,129:881-890
11LING Guan-Ting.Cereals&Oils.,2009,3:41-43凌關(guān)庭.糧食與油脂,2009,3:41-43
12YANG Qing.China Quality Supervision.,2010,5:35-37楊晴.中國(guó)質(zhì)量技術(shù)監(jiān)督,2010,5:35-37
13Prosky L,Dell R G.J.Pharm.Sci.,1971,60:1341-1343
14Renwick A G,Williams R T.J.Biochem.,1972,129:857-867
15Matsumura R,Kohei H.J.Chromatogr.A,1976,117:383-391
16Isidre C,Maria R,Judith A,Margarita F,Lluis SM.J.Chromatogr.A,1996,750:397-402
Ultra Liquid Chromatography-Tandem Mass Spectrometry with Dansyl Chloride Pre-column Derivatization
YANG Hua-Mei*,HANG Li,YANG Jian-Guo,ZHANG Xue-Mei,HUANGWei-Hong,LIChen,JIANG Xiao-Li
(Taizhou Center for Disease Control and Prevention,Taizhou 225300,China)
A method was developed for the quantification of cyclohexylamine in human urine samples by ultra performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry(UPLCESI-MS/MS)withpre-columnderivatization.After the refrigeratedcentrifugationof urine samples,cyclohexylamine in supernatantwas derived with dansyl chloride and then its derivant was purified with solid phase extraction(SPE).The target compounds were separated on a Waters ACQUITY CSHTM C18column (50 mm×2.1 mm,1.7μm)with the gradient elution of methanol and water(containing 0.002 mol/L ammonium acetate)and were determined in electrospray positive ionization(ESI+)and multiple reaction monitoring(MRM)modes.The linear range was 2.5-200μg/L for cyclohexylamine with the correlation coefficientmore than 0.999.The recovery ranged from 98.7%to 102.3%,with the relative standard deviations (RSDs)of 3.1%-5.2%.The limit of detection(LOD)and the limit of quantitation(LOQ)were 1μg/L and 3μg/L.The results indicated that the method was accurate and reliable,and suitable for the detection of cyclohexylamine in urine samples.The urine samples of 200 primary school students were analyzed by this method and the positive rate of cyclohexylamine was 34.5%.
Cyclohexylamine;Dansyl chloride;Derivatization;Solid phase extraction;Human urine samples;Ultra liquid chromatography-tandem mass spectrometry
28 September 2015;accepted 7 December 2015)
10.11895/j.issn.0253-3820.150764
2015-09-28收稿;2015-12-07接受
*E-mail:yanghuamei263@sina.com
