基于高通量測序技術的實驗動物沙門氏菌檢測方法的建立與評價
2016-11-29 08:45:10胡毅翔張歡歡余陳歡金曉音應華忠
中國比較醫學雜志 2016年10期
胡毅翔,張歡歡,余陳歡,金曉音,應華忠
(浙江省醫學科學院 浙江省實驗動物與安全性研究重點實驗室, 杭州 310013)
?
技術方法
基于高通量測序技術的實驗動物沙門氏菌檢測方法的建立與評價
胡毅翔,張歡歡,余陳歡,金曉音,應華忠
(浙江省醫學科學院 浙江省實驗動物與安全性研究重點實驗室, 杭州 310013)
目的 建立基于高通量測序技術的實驗動物沙門氏菌檢測方法,應用于實驗動物沙門氏菌的檢測。方法 提取小鼠糞便DNA樣本,分別針對16S rDNA區域、23S rDNA區域、16S~23S rDNA IS、23S~5S rDNA IS、gyrB優選區域設計通用引物,對各個引物進行測試分析,優化擴增條件并建庫,利用Illumina高通量測序技術檢測區分42個樣本中的沙門氏菌,評價該方法的特異性和穩定性。結果 篩選發現沙門氏菌的菌種分類優選區域為gyrB基因,gyrB基因引物序列為FP5’-AACCACCGCAATCAGACCTT3’,FP5’-AGCCACGAAACCTTCACYA-3’。對引物進行優化,確定最佳擴增條件及樣品上樣量并正式建庫,通過高通量測序和序列分析能檢測出42個樣品中極微量的沙門氏菌,檢測方法穩定,靈敏度高,檢測限可達0~102的cfu。結論 本實驗利用高通量測序技術,建立了一套完整檢測實驗動物沙門氏菌的生物體系,能檢測出實驗動物體內極微量的沙門氏菌,檢測方法穩定性好,靈敏度高,可沿用至其他種類病原微生物的檢測。
沙門氏菌;實驗動物;高通量測序
沙門氏菌(Salmonellae)是一類可以引起沙門氏菌病的典型人畜共患病原菌,屬腸桿菌科 (Enterobacteriaceae),兼性厭氧型革蘭氏陰性菌[1,2]。自1885年Salmon等分離得到豬霍亂沙門氏菌以來,目前已檢出沙門氏菌血清型約2 500余種,其中我國發現的100多種[3]。沙門氏菌在分類學上被分為兩個種,分別為Salmonellaenterica和Salmonellabongori,可由多種途徑向人類進行傳播,進入人體后在腸道內寄生并繁殖,所產生的沙門氏菌內毒素能進入血液循環,引發腸道黏膜腫脹、脫落、甚至潰瘍、壞死[4]。在實驗室研究方面,攜帶或者潛在攜帶沙門氏菌的實驗動物不僅對實驗人員造成潛在的傳染隱患,也會對后續科學實驗產生較大的影響,因此對于實驗動物的沙門氏菌的檢測尤為重要[5]。
生化方法和免疫學方法是目前檢測沙門氏菌的主要方法,但這類方法檢驗程序復雜繁瑣,耗時長,部分檢驗過程及結果判定存在實驗人員的主觀判斷,因此新的檢測方法的引入勢在必行[6]。分子生物學方法具有快速、準確的優勢,現已開始被大量的應用于檢測病原微生物,主要涉及到的分子生物學技術主要包括PCR技術、基因芯片和環介導等溫擴增技術(LAMP)等[7,8]。近年來,高通量測序技術開始引入病原微生物檢測領域,實現了更為高效的檢測手段。本次研究主要采用的是Illumina的Miseq高通量測序平臺,對于病原微生物檢測該平臺具有周期短、通量高、成本低、準確率高等優點[9,10]。本實驗以分子生物學技術方法為基礎,采用Miseq高通量測序技術檢測實驗室用小鼠所攜帶的沙門氏菌,通過本次研究,建立了一套完整檢測實驗動物沙門氏菌的技術體系。
1 材料和方法
1.1 動物
SPF級ICR小鼠42只,體重18~20 g,由浙江省醫學科學院實驗動物中心提供 (實驗動物生產許可證號為 SCXK(浙) 2008-0034,使用許可證號為SYXK (浙) 2008-0114。
1.2 菌株
鼠傷寒沙門氏菌凍干株(編號: TA98),甲型副傷寒沙門氏菌凍干株(編號: CMCC(B)50093),腸炎沙門氏菌凍干株(編號: TDF918),志賀菌凍干株(編號: TG212)為本實驗室冷凍保存。
1.3 藥品和試劑
PremixExTaqTMHot Start Version(日本Takara公司,批號RR030A),DL2000 DNA Marker(日本Takara公司,批號3427),Axygen PCR Clean Up Kit(美國Axygen公司,批號AP-PCR-500),QubitdsDNA HS Assay Kit(美國Invitrogen公司,批號Q32851),PrimeScript? RT reagent Kit(日本Takara公司, 批號DRR037A)。高通量測序試劑盒(Illumina,美國),Trizol reagent(Invitrogen,美國,批號Q35438)。
1.4 主要儀器
Bioanalyzer 2100 生物分析儀(Agilent,美國);Hiseq 2500 高通量測序儀(Illumina,美國);熒光定量 PCR 儀(Bio-Rad,美國);Qubit 2.0核酸定量儀(Life Technologies,美國);組織研磨儀(TissueLyser,Qiagen,德國)。
1.5 實驗方法
1.5.1 小鼠沙門氏菌模型的建立及樣品收集: 小鼠適應性飼養3 d后,隨機分為6組: 鼠傷寒沙門氏菌組(5只)、甲型副傷寒沙門氏菌組(5只)、志賀桿菌組(5只)、腸炎沙門氏菌和甲型副傷寒沙門氏菌(5只)、腸炎沙門氏菌組(18只)、陰性對照組(4只)。鼠傷寒沙門氏菌組每只小鼠灌服鼠傷寒沙門氏菌,甲型副傷寒沙門氏菌組每只小鼠灌服鼠傷寒沙門氏菌0.2 mL (5×103cfu/ mL),志賀桿菌組每只小鼠灌服鼠傷寒沙門氏菌0.2 mL (5×103cfu/ mL),腸炎沙門氏菌和甲型副傷寒沙門氏菌每只小鼠灌服腸炎沙門氏菌和甲型副傷寒沙門氏菌0.2 mL (5×103cfu/ mL),腸炎沙門氏菌組每只小鼠灌服鼠傷寒沙門氏菌0.2 mL(0 cfu/mL、5×101cfu/mL、5×102cfu/mL、5×103cfu/mL、5×104cfu/mL、5×105cfu/mL),每個濃度各3只,各組小鼠每隔2周灌胃1次,連續3次。取60 mg小鼠糞樣置于2 mL離心管中,采用PowerFecal DNA Isolation Kit 試劑盒(MoBio,美國) 抽提100 mg小鼠糞便樣本中微生物的總 DNA, 操作步驟按照說明書進行,完成基因組DNA抽提后,放-20℃保存備用,樣品信息見表1。

表1 樣品信息
1.5.2 菌種分類優選區域選擇: 通過文獻查找,針對沙門氏菌,尋找在研究范圍內的幾個菌種相互之間差異變化大的區域。NCBI數據庫中查找沙門氏菌典型菌株,以鼠傷寒沙門氏菌(Salmonellaentericaserovar Typhimurium)典型菌株LT2、ST4/74等作為研究對象。通過序列比對,去除經比對菌種間同源關系極高的區域,最終篩選得到5大類區域: ①16S rDNA區域,②23S rDNA區域,③16S~23S rDNA ISR,④23S~5S rDNA ISR,⑤gyrB優選區域。
1.5.3 通用引物設計及優化: 針對①16S rDNA區域,②23S rDNA區域③16S~23S rDNA ISR,④23S~5S rDNA ISR,⑤gyrB優選區域設計通用引物,通用引物在多個菌種中都能PCR擴增得到產物。對設計的引物進行引物測試,淘汰不能擴增的引物。在原有的通用引物基礎上加上建庫和測序需用到的接頭序列(Linker),區分樣本使用的正反向Barcode序列(Index),增加終文庫均一性和豐富度的連接堿基(heterogeneity spacer),如此設計的引物長度在90 bp~97 bp之間,該優化引物可直接用于建庫,實際的建庫工作需要一步PCR反應完成。優化引物設計圖如圖1所示:

圖1 優化引物結構組成圖Fig.1 Diagram of optimization of the primer structure
1.5.4 沙門氏菌建庫PCR條件摸索: 建庫PCR采用的是90~97 bp長度的長鏈引物,這與普通16~25 bp的引物PCR擴增條件會有很多不同,長鏈引物擴增條件比普通引物擴增的條件要苛刻。建庫PCR條件摸索: 模板上樣量、退火溫度、循環、延伸時間,其中主要考查上樣量和退火溫度。(1)退火溫度: 考察的退火溫度為56℃、54℃、52℃。2) 模板上樣量: 針對模板上樣量設計11個模板濃度梯度實驗,模板信息如表1所示。上樣量共分為12個梯度,依次為500、200、100、50、25、10、1 ng、100、10、1,100,10 fg。
1.5.5 高通量測序體系的特異性和敏感性檢測: 42個檢測樣本的建庫后采用 Illumina Miseq 高通量測序平臺進行高通量測序。隨后對高質量序列進行提取, 提取方法: Miseq 測序得到的 PE reads 首先根據 overla關系進行拼接, 將成對的 reads 拼接成一條序列, 同時對reads 質量和 merge 的效果進行質量質控和過濾, 去除序列末端的后引物和接頭序列、多堿基 N、polyA/T尾巴及低質量堿基; 去除所得序列的 barcode 標簽序列、前引物序列;丟棄長度短于 200 bp、模糊堿基數>0、序列平均質量低于20的序列。高通量測序結果與樣本信息進行對比,評價高通量測序體系檢測沙門氏菌的特異性和敏感性。
2 結果
2.1 通用引物設計及引物測試
針對①16S rDNA區域,②23S rDNA區域, ③16S~23S rDNA ISR,④23S~5S rDNA ISR,⑤gyrB優選區域各設計3對通用引物。經過測試,16S rDNA、23S rDNA、gyrB各有一對引物通過測試。但分析后發現16S rDNA、23S rDNA的引物實際建庫測序的結果與設計時的有偏差,它無法區分樣品中的腸炎沙門氏菌。最后確定沙門氏菌的菌種分類優選區域為gyrB基因引物(表2)。gyrB基因通用引物引物測試電泳結果如圖2所示,由于gyrB基因不僅存在于沙門氏菌中,在志賀桿菌、大腸桿菌等腸科桿菌中都存在該基因,因此樣品中只要有以上任一菌種,都能擴增出目的條帶。引物主要是優勢擴增沙門氏菌,但是該引物對于其他沙門氏菌和腸科桿菌的模板DNA還是有微弱的結合能力,因此即使是沙門氏菌陰性樣品,也能夠擴增得到目的條帶。針對gyrB基因設計的1對引物(測試了鼠傷寒沙門氏菌和志賀桿菌其中的兩個樣本),條帶單一,條帶大小約為456 bp,通過電泳圖可以看到此對引物對沙門菌的gyrB基因優勢擴增,條帶較亮,對屬于腸桿菌屬的志賀桿菌的gyrB基因擴增能力有很大程度降低,條帶較淡。

表2 gyrB基因通用引物基本信息
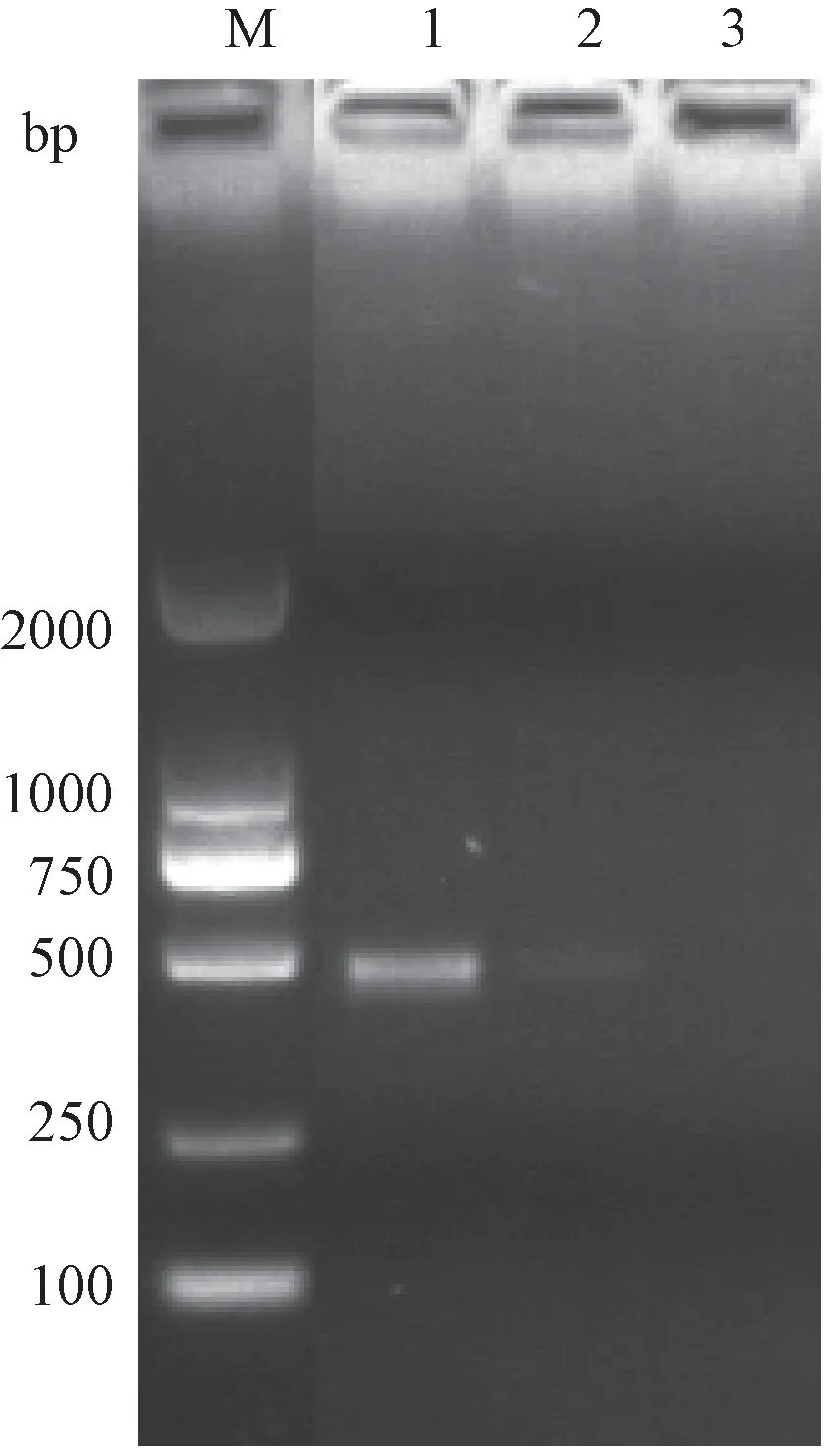
M-DL2000 marker 1-鼠傷寒沙門氏菌樣本 2-志賀桿菌樣本 3-無模板陰性對照圖2 gyrB基因通用引物引物測試電泳圖M-DL2000 marker. 1: Salmonella typhimurium sample; 2: Shigella sample; 3: Negative control.Fig.2 Electrophoregram of the universal primers for GyrB gene
2.2 沙門氏菌建庫PCR條件優化
2.2.1 退火溫度: 第1輪PCR,退火溫度56℃,無擴增條帶,未成功;第2輪PCR,退火溫度54℃,無擴增條帶,未成功;第3輪PCR,退火溫度52℃,有擴增條帶,成功。

M: DL2000 marker模板量為1: 500 ng;2: 200 ng;3: 100 ng;4: 50 ng;5: 25 ng;6: 10 ng;7: 1 ng;8: 100 pg;9: 10 pg;10: 1 pg;11: 100 fg;12: 10 fg。圖3 沙門氏菌模板濃度梯度實驗電泳結果圖M: DL2000 Marker. Loading quantity of samples. 1: 500 ng; 2: 200 ng; 3: 100 ng; 4: 50 ng; 5: 25 ng; 6: 10 ng; 7: 1 ng; 8: 100 pg; 9: 10 pg; 10: 1 pg; 11: 100 fg; 12: 10 fg.Fig.3 Results of concentration gradient experiment of the Salmonella templates
2.2.2 模板上樣量: 針對模板上樣量設計一個模板濃度梯度實驗,共分為12個梯度,依次為500、200、100、50、25、10、1 ng、100、10、1、100、10 fg,實驗結果如下電泳圖3。通過以上模板濃度梯度實驗,可以發現模板在10 ng~200 ng范圍內,都能擴增得到目的片段。為了保證最后的產物有足夠的量,也就是要保證目的條帶足夠亮,選擇較為合適的模板濃度150 ng作為體系最終的模板濃度。總結沙門氏菌gyrB基因建庫PCR體系為DNA 150 ng、PremixExTaq12.5 μL,上下游優化引物 ( 10 pmol /μL) 各2.5 μL,補充ddH2O至20 μL;PCR最佳反應條件為: 98℃ 預變性 30 s; 98℃ 變性 10 s,52℃退火30s, 72℃延伸 10 min,共 40個循環; 72℃再延伸 5 min。
2.3 樣本正式建庫
采用以上摸索得到的建庫PCR條件將所有樣品進行建庫,建庫PCR結果如圖4~6所示。結果顯示,42個樣本建庫成功,均可得到456 bp大小的目的條帶。
2.4 檢測結果的準確性、特異性及重復性分析
42個樣本的高通量測序檢測結果如表3所示,各樣本檢測到的主體菌種與目的菌種基本上一致的。通過測序的方法在樣品中存在的極微量的菌種也被檢測到,因此在一些樣品中除了要檢測的主體菌種外,還存在極微量拷貝數的其他沙門氏菌菌種。在志賀桿菌中,特別設置了一個志賀桿菌的數據庫用于19~23號樣品的比對,發現無法檢測到該菌種,說明設計的引物可以特異擴增沙門氏菌。在混合菌種中,該引物能將兩種不同的菌種進行區分。針對腸炎沙門氏菌設置的cfu梯度,從檢測的結果可以看出,在0~102的cfu范圍內,也能檢出,在其他cfu梯度樣品中都能穩定檢出,準確性及靈敏度高,各平行樣本之間的檢測結果一致,說明該方法具有較高重復性。通過高通量測序技術能夠將檢測出極微量的沙門氏菌,并能沙門氏菌很好的區分出來。

M: DL2000 marker 1-17 :1-17號樣本圖4 1-17號樣本建庫PCR電泳圖M: DL2000 marker 1-17 : Sample 1-17Fig.4 PCR electrophoregram of the sequencing library of samples 1-17

M: DL2000 marker. 18-34:18-34號樣本圖5 18-34號樣本建庫PCR電泳圖M: DL2000 marker. 18-34: Samples 18-34Fig.5 PCR electrophoregram of the sequencing library of samples 18-34

表3 沙門氏菌樣本的數據分析結果
注: ST:Salmonellaentericaserovar Typhimurium;鼠傷寒沙門氏菌;SA:Salmonellaentericaserovar Paratyphi A;甲型副傷寒沙門氏菌;SE:Salmonellaentericaserovar Enteritidis;腸炎沙門氏菌;SH: Shigella;志賀菌

M: DL2000 marker. 35-42: 35-42號樣本圖6 35-42號樣本建庫PCR電泳圖M: DL2000 marker. 35-42: Sample 35-42Fig.6 PCR electrophoregram of the sequencing library of samples 35-42
3 討論
人畜共患病是指由同一種病原體引起,流行病學上相互關聯,在人類和動物之間自然傳播的疫病。其病原包括病毒、細菌、支原體、螺旋體、立克次氏體、衣原體、真菌、寄生蟲等。這類疾病的特性在于很不容易消滅。因此這些病原微生物的存在會對人類健康、畜牧業安全生產、畜產品安全和公共衛生造成重大危害[11]。沙門氏菌是一類可以引起沙門氏菌病的典型人畜共患病原菌, 并且在實驗動物中十分常見,可感染實驗人員并對實驗結果造成不可預估的影響,因此對于沙門氏菌的檢測就變得尤為重要。
如今,分子生物學技術已越來越多的被應用于菌種鑒定,比如現在較為成熟的16S rDNA細菌菌種鑒定技術,ITS區真菌菌種鑒定技術等等,都體現出了簡便、快速、高效的特點。菌種鑒定到屬的級別還是相對較容易的,現在也有很多技術可以達成,但是想要鑒定到種、亞種甚至是菌株的水平,鑒定的困難程度就要提高很多。16S rDNA能很好的將菌種鑒定到屬的層面, 但是由于16S rDNA的進化速度非常慢,即太保守,雖對種系有一定的區分能力,但是難以區分某些種系非常接近的菌種及同一菌種的不同菌株。23S rDNA也可用于菌種鑒定,有些學者認為其系統進化分析能力甚至比16S rDNA更強,但是由于23S rDNA的分子大小較大,因此它的使用也受到了限制[12,13]。核糖體基因間隔區(intergenic spacer region, ISR)為rDNA區域之間高度可變的區域,它的進化速度遠遠的高于rDNA區域,可以用于菌種間的鑒別。ISR區域主要有16S ~23S rDNA ISR區域和23S~5S rDNA ISR區域。ISR區域堿基變化程度與rDNA區域的程度相差不多,它優于rDNA區域的原因主要是該區域存在片段插入或缺失,導致片段大小的變化從而產生了種間和種內差異[14]。Morales等還發現23S~5S rDNA ISR區域比16S ~23S rDNA ISR區域具有更好的菌種區分能力[15]。然而,在某種程度上,ISR區域的種系鑒別能力也因其高度變異性和高度進化率而受到限制。
細菌中的一些基因也可以作為系統發育標記基因,比如gyrB基因,該基因編碼DNA解旋酶(gyrase)的B亞單位,具有DNA依賴的ATP酶活性,催化ATP的水解。Fukushima應用gyrB基因對大腸埃希菌、沙門氏菌和志賀桿菌進行系統發育分析,證實了gyrB基因優于16S rDNA基因[14]。由于gyrB基因的進化速度比16S rDNA等其他基因快,因此更適用于親緣關系較近的種內及種間的鑒定和分類。
在本研究中,針對16S rDNA、3S rDN、16S~23S rDNA IS、23S~5S rDNA IS、gyrB優選區域設計通用引物,對各個引物進行測試分析, 最終篩選發現沙門氏菌的菌種分類優選區域為gyrB基因,gyrB基因引物序列FP 5’-AACCACCGCAATCAGACCTT-3’,RP 5’-AGCCACGAAACCTTCACYA-3’。優化擴增條件并建庫,利用Illumina高通量測序技術檢測區分42個樣本進行通過高通量測序和序列分析,能夠檢測出樣本中極微量的沙門氏菌,檢測方法穩定,檢測限可達0~102的cfu,檢測方法穩定性好,靈敏度高,可沿用至其他科學研究用的動物(如大鼠、豚鼠、家兔等)病原微生物的檢測,同時還可將該實驗體系應用于其他種類病原微生物的檢測。
[1] 王軍, 鄭增忍, 王晶鈺. 動物源性食品中沙門氏茵的風險評估 [J]. 中國動物檢疫, 2007, 24(4): 23-25.
[2] 張嘉寧, 藏富妍, 顧為望. 應用PCR法檢測小鼠沙門氏菌 [J]. 中國比較醫學雜志, 1999, 9(1): 45-47.
[3] McGuinness S, McCabe E, O’Regan E, et al. Development and validation of a rapid real-time PCR based method for the specific detection of Salmonella on fresh meat [J]. Meat Sci, 2009, 83(3): 555-562.
[4] Liu FP, Tong HM.GyrAandParCgene mutation of clinically isolated fluoroquinolone-resistant strain of salmonella [J]. J Northeast Agr Univ, 2006,13(1): 47-50.
[5] Yan SS, Pendrak ML, Abela-Ridder B, et al. An overview of Salmonella typing — public health perspectives [J]. Clin Appl Immunol Rev, 2003, 4(3): 189-204.
[6] 范媛媛, 鄭冰, 王樹祥, 等. 一種新型培養基在沙門氏菌檢測中的效果評價 [J]. 食品工業科技, 2008, 8: 415-417.
[7] 楊保偉, 盛敏, 席美麗, 等. 食源性沙門氏菌耐藥性檢測及相關質粒 [J]. 微生物學報, 2008, 48(8): 1006-1012.
[8] He LL, Sok D, Azadnia P, et al. Toward a more accurate view of human B-cell repertoire by next-generation sequencing, unbiased repertoire capture and single-molecule barcoding [J].Sci Rep, 2014, 4: 67-78.
[9] Weinstein JA, Jiang N, White III RA, et al. High-throughput sequencing of the Zebrafish antibody repertoire [J]. Science, 2009, 324(5928): 807-810.
[10] DeKosky BJ, Ippolito GC, Deschner RP, et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire [J]. Nat Biotech, 2013, 31(2):166-169.
[11] 王章云, 滕煥昭, 李柏桂, 等. 腸炎沙門氏菌引起的食物中毒細菌學調查 [J]. 中國人獸共患病雜志, 1999, 15(3): 115.
[12] Henrik C, Steen N, John EO. Phylogenetic relationships of Salmonella based on rRNA sequences [J]. Int J Systematic Bacteriol, 1998, 48: 605-610.
[13] Sara PL, Francisco R, Ruiting L. Variation of the ribosomal operon 16S-23S gene spacer region in representatives ofSalmonellaentericasubspecies [J]. J Bacteriol, 1998, 180(8) :2144-2151.
[14] Cesar AM, Richard G, Jean G. Linkage of avian and reproductive tract tropism with sequence divergence adjacent to the 5S ribosomal subunitrrfHofSalmonellaenterica[J]. FEMS Microbiol Lett, 2006, 264: 48-58.
[15] Fukushima M, Kakinuma R. Phylogenetic analysis of Salmonella, Shigella, and Escherichia coli strains on the basis of thegyrBgene sequence [J]. J Clin Microbiol, 2002, 40(8): 2779-2785.
Establishment and evaluation of a high throughput sequencing technology for detection ofSalmonellain laboratory animals
HU Yi-xiang, ZHANG Huan-huan, YU Chen-huan, JIN Xiao-yin, YING Hua-zhong
(Zhejiang Key Laboratory of Experimental Animal and Safety Evaluation, Zhejiang Academy of Medical Sciences, Hangzhou 310013, China)
Objective To establish a detection method ofSalmonellain laboratory animals based on a high-throughput sequencing technology, and to apply it in detection ofSalmonellain laboratory animals. Methods DNA samples were extracted from mouse feces. Universal primers for 16S rDNA, 23S rDNA, 16S-23S rDNA, 23S-5S rDNA region, gyrB preferred area were designed, respectively. Each primer was tested and analyzed to determine the best amplification conditions and build a database. Forty-two samples ofSalmonellawere assayed by Illumina high-throughput sequencing technology and evaluated the specificity and stability of this method. Results The species preferred region ofSalmonellawas gyrB gene region. The primers for gyrB gene were FP5’-AACCACCGCAATCAGACCTT3‘ and FP5’-AGCCACGAAACCTTCACYA-3’. The primers were optimized and determined, through a high-throughput sequencing, and the sequence analysis detected very small amount ofSalmonellain the 42 samples, indicating that this detection method is stable, highly sensitive, and the limit of detection reached to 0-102CFU. Conclusions We have established a complete detection system for detection ofSalmonellain laboratory animals based on a high-throughput sequencing technology, This system can detect trace amounts ofSalmonellain laboratory animals, and this detection method is stable and highly sensitive, which can be also used in detection of other kinds of pathogenic microorganism in laboratory animals.
Salmonella; Laboratory animal; High-throughput sequencing
浙江省衛生高層次人才(2012F30026),浙江省科技廳院所專項(2016F30001)。
胡毅翔(1989-),男,碩士研究生,研究方向為中藥藥理學。Email: aahyxaa@163.com。
應華忠(1968-),男,研究員,從事動物模型與比較醫學研究。 Email: YHZ0101@126.com。
R-33
A
1671-7856(2016)10-0072-07
10.3969.j.issn.1671-7856.2016.10.014
2016-03-08
猜你喜歡
今日農業(2021年9期)2021-11-26 07:41:24
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
發明與創新·小學生(2021年3期)2021-03-25 11:48:49
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
海峽科技與產業(2016年3期)2016-05-17 04:32:12
中國科技博覽(2016年2期)2016-04-25 20:32:39
小學生導刊(2016年34期)2016-04-11 00:49:44
