Peters綜合征臨床特征和基因突變的研究
2016-12-05 08:58:59黃麗琴盧國(guó)華毛平安孫新成
國(guó)際眼科雜志 2016年12期
關(guān)鍵詞:基因突變
黃麗琴,盧國(guó)華,謝 陽(yáng),毛平安,孫新成,孟 永
?
·臨床研究·
Peters綜合征臨床特征和基因突變的研究
黃麗琴1,盧國(guó)華1,謝 陽(yáng)1,毛平安1,孫新成1,孟 永2
?METHODS: Ten congenital corneal opacities affected patients were enrolled from our pediatric and genetic eye clinic. Medical and ophthalmic histories were obtained. Genomic DNA was prepared from venous leukocytes after informed consent conforming was obtained from each participant. The coding regions and the flanking exon-intron junctions of this gene were amplified by polymerase-chain reaction (PCR) and subsequently analyzed by direct sequencing. Variations detected were further evaluated in 100 normal controls by direct sequencing.
?RESULTS: One affected individual characterized by systemic changes such as congenital heart anomalies and hearing loss showed the consistent phenotypes with Peters syndrome. Sequence analysis of the PITX2 gene revealed one novel mutation, c. 788G>A. Nucleotide sequence analysis showed that this mutation led to the functional abnormal of this gene, however, no mutation was observed in any unaffected member or 100 normal unrelated individuals.
?CONCLUSION: A novel mutation in the PITX2 gene have been identified, this is the first report on a mutation in a Chinese Peters syndrome and the result expand the mutation spectrum of PITX2, further clarify the clinical features of the disease. All these will be useful foundations for clinical diagnosis, medical therapy and pathogenesis.
目的:分析國(guó)人Peters綜合征的臨床特征,并確定致病基因突變,為該病的臨床診斷和治療及發(fā)病原因提供依據(jù)。
方法:選取2012/2015年在常州市第二人民醫(yī)院眼科就診的10例先天性角膜混濁的患者,并收集詳細(xì)的相關(guān)臨床資料。征得患者及其家系成員的同意后抽血制備基因組DNA,用聚合酶鏈反應(yīng)(PCR)對(duì)致病基因PITX2的編碼區(qū)及其臨接內(nèi)含子進(jìn)行擴(kuò)增后,直接測(cè)序分析該基因。同時(shí)檢測(cè)100位無(wú)親緣關(guān)系的正常人外周血標(biāo)本進(jìn)行對(duì)照驗(yàn)證。
結(jié)果:患者1例的臨床特點(diǎn)包括先天性角膜中央部混濁白斑,伴有相應(yīng)區(qū)域的角膜后部基質(zhì)變薄和后彈力層缺損,且患者伴有全身系統(tǒng)如心臟和聽力損害等改變,符合Peters綜合征的臨床診斷;該患者PITX2基因突變篩查結(jié)果發(fā)現(xiàn)了1種新突變,c. 788G>A,導(dǎo)致該基因的功能異常,而家屬中表型正常者及無(wú)親緣關(guān)系的正常對(duì)照者均未發(fā)現(xiàn)該基因突變。
結(jié)論:先天性角膜混濁患者10例中檢測(cè)到1個(gè)新PITX2基因突變,符合Peters綜合征的臨床診斷,這是中國(guó)首次報(bào)道Peters綜合征的PITX2基因突變,結(jié)果豐富了PITX2基因突變頻譜,并進(jìn)一步明確了Peters綜合征的臨床特點(diǎn),為該病的臨床診斷和治療及發(fā)病原因提供了依據(jù)。
Peters綜合征;先天性角膜混濁;PITX2基因;突變
引用:黃麗琴,盧國(guó)華,謝陽(yáng),等.Peters綜合征臨床特征和基因突變的研究.國(guó)際眼科雜志2016;16(12):2237-2240
0引言
Peters綜合征(簡(jiǎn)稱PPS)最初在20世紀(jì)由Peter依據(jù)臨床表現(xiàn)及組織學(xué)特點(diǎn)進(jìn)行描述,是最常見的一種先天性角膜混濁眼病。國(guó)外報(bào)道在新生兒中的發(fā)病率為1.2/100 000,其大多數(shù)是散發(fā)病例,但也有一些染色體顯性及隱性遺傳的報(bào)道。PPS是臨床中少見病,且易與其他引起先天性角膜混濁的眼病如鞏膜化角膜、角膜皮樣瘤、先天性青光眼、小眼畸形、產(chǎn)傷性角膜白斑及黏多糖貯積癥眼病等相混淆。國(guó)外已有較多關(guān)于本病分子遺傳學(xué)的研究,在國(guó)內(nèi),目前尚缺少該眼病的的致病基因的研究及臨床特征的描述,以致很多臨床醫(yī)師沒有足夠認(rèn)識(shí)。本研究收集10例先天性角膜混濁病患者的臨床資料,明確PPS的臨床特點(diǎn),利用PCR和直接測(cè)序方法對(duì)其致病基因PITX2進(jìn)行突變檢測(cè),了解PPS基因型與表型的關(guān)系,為該病的臨床診斷和治療及發(fā)病原因提供依據(jù)。
表1 PCR反應(yīng)所需引物的序列和相應(yīng)擴(kuò)增產(chǎn)物的長(zhǎng)度與退火溫度

基因外顯子正向引物(5 -3 )反向引物(5 -3 )產(chǎn)物長(zhǎng)度(bp)退火溫度(℃)PITX21GGCCGCCGCTTCTTACACACTGGCGATTTGGTTCTGATTT254632CTGCCTCGCCCCTCCCCCTCCTCTTCAAGCAGCAGGCGCGTCAGGTC350643TCTTCGTTTGCCCGCTGACTTTGGCGAACGACCACTCCCACCAC549634GGCGCGGACACCCACACTTGGCGCTGCCTTCCACATTCTCT487655-1CTGGCCCTGGTATCTTGGTGTGCGGTGGATAGGGAGGCGGATGTAAG346635-2CGACGACATGTACCCAGGCTATTCCGACGGGCTACTCAGGTTGTTCA256645-3TCCCGGGCTCCAGTCTCAACAGTTTCTTTAGTGCCCACGACCTTCT35564
注:外顯子5共設(shè)計(jì)三對(duì)引物進(jìn)行PCR擴(kuò)增;PCR反應(yīng)體系中PITX2的GC含量較高,故引物采用2×GC Buffer I(Takara)的緩沖液。
1對(duì)象和方法
1.1對(duì)象 選取先天性角膜混濁眼病臨床標(biāo)準(zhǔn):(1)先天性角膜混濁白斑;(2)患眼角膜無(wú)血管化;(3)可伴有其它眼部異常:包括小角膜、小眼球、鞏膜化角膜、中央凹發(fā)育不良、視乳頭發(fā)育不良、青光眼、白內(nèi)障、永存原始玻璃體增生癥等;(4)可伴有全身異常:身材短小、聽力損害、心臟疾病等。符合以上標(biāo)準(zhǔn)的共有10個(gè)沒有親緣關(guān)系的初步診斷為先天性角膜混濁的患者參加本項(xiàng)研究,詳細(xì)詢問所有受試者病史,并行眼部檢查以及全身檢查。遵循相關(guān)倫理原則,征得患者及其家系成員的同意后抽血制備基因組DNA,并收集相關(guān)臨床資料。100例正常對(duì)照者均無(wú)已知眼遺傳病及全身遺傳病,與患者無(wú)親緣關(guān)系。
1.2方法
1.2.1聚合酶鏈反應(yīng)擴(kuò)增 采集10例先天性角膜混濁患者肘靜脈血5mL,用常規(guī)酚氯仿方法,從患者和100例正常人外周血中提取DNA。根據(jù)Genbank中人PITX2基因cDNA序列,采用Primer 5.0軟件設(shè)計(jì)引物(表1)。采用聚合酶鏈反應(yīng)(PCR)擴(kuò)增PITX2基因所有外顯子和臨近內(nèi)含子。PCR反應(yīng)體系為20μL,包含模板DNA 40ng/μL,2×GC緩沖液12.5μL,引物(上海生工公司)0.4μL×2,2.5mmol/L dNTP 0.6μL,rTaq酶0.4μL。PCR反應(yīng)條件:94℃預(yù)變性5min,94℃變性30s,63℃~65℃退火30s,72℃延伸30s,30~34個(gè)循環(huán),反應(yīng)終止后72℃后延伸5min,4℃保存。瓊脂糖電泳:應(yīng)用15g/L瓊脂糖凝膠水平電泳檢測(cè)PCR產(chǎn)物,在紫外燈下觀察是否有電泳條帶、產(chǎn)物量多少和是否有非特異性擴(kuò)增。
1.2.2突變基因篩查 對(duì)PCR產(chǎn)物純化,使用AB13730 DNA全自動(dòng)測(cè)序儀上機(jī)進(jìn)行雙向測(cè)序,測(cè)序結(jié)果采用Lasegen軟件進(jìn)行對(duì)比分析,結(jié)果與Genbank中人類基因的標(biāo)準(zhǔn)系列進(jìn)行比對(duì),測(cè)序識(shí)別的PITX2新突變與100位無(wú)親緣關(guān)系的正常人進(jìn)行測(cè)序?qū)Ρ确治觥?/p>
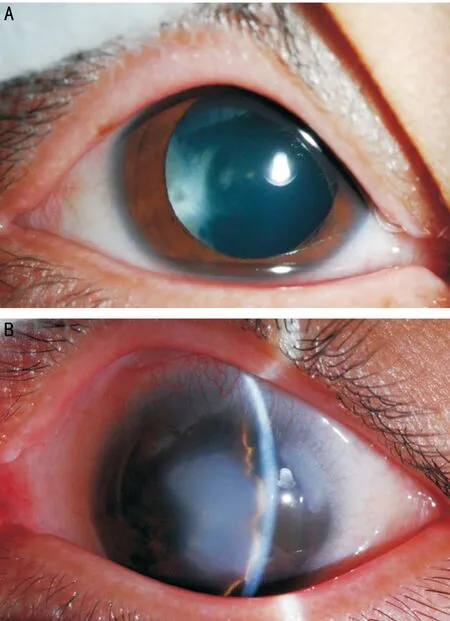
圖1 突變患者雙眼眼前節(jié)照相 A:右眼;B:左眼。
1.2.3 PITX2蛋白的生物信息學(xué)分析 從ensembl數(shù)據(jù)庫(kù)中下載人類PITX2分子的氨基酸序列,采用在線工具(http://www.sbg.bio.ic.ac.uk/)中的Phyre2軟件分析突變前后的PITX2蛋白質(zhì)的三級(jí)結(jié)構(gòu)。對(duì)突變位點(diǎn)可能導(dǎo)致的氨基酸變化的差異以及可能致病性通過PolyPhen軟件進(jìn)行評(píng)估。
2結(jié)果
在選取的10例先天性角膜混濁患者中,檢測(cè)到1個(gè)新PITX2基因突變,分析其詳細(xì)的相關(guān)臨床資料,符合Peters綜合征的臨床診斷,這是中國(guó)首次報(bào)道Peters綜合征的PITX2基因突變。
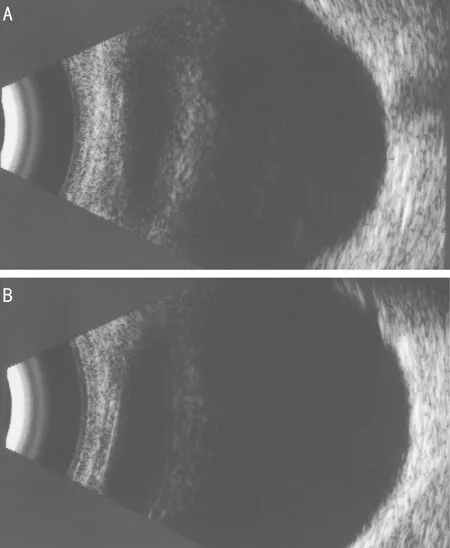
2.1 PITX2突變患者病例資料 患者,男,8歲,因雙眼脹痛伴視物模糊1mo就診。既往史:患者父母訴患兒出生后即被發(fā)現(xiàn)左眼角膜發(fā)白(患者雙眼眼前節(jié)照相見圖1)。4歲因患 “室間隔缺損”行室間隔缺損修補(bǔ)手術(shù)。7歲時(shí)被診斷出雙耳聽力下降。眼科檢查:VOD:0.15(-1.00DS矯正0.2),VOS:0.1。右眼:角膜透明,前房深度可,晶狀體白色混濁,眼底窺不清;左眼角膜中央變薄、白色圓形混濁大小約5mm,后彈力層變薄,前房軸深2CT,房角4∶00~6∶00角膜緣后可見后胚胎環(huán),瞳孔圓,對(duì)光反射正常,散瞳后見晶狀體透明,眼底檢查窺不清;患兒A超檢查雙眼眼圖2 突變患者眼部B超:雙眼后節(jié)(-) A:右眼;B:左眼。
軸:OD:21.02mm,OS:21.00mm;雙眼眼部B超:雙眼后節(jié)(-)(圖2)。房角鏡檢查:雙眼房角粘連大于1/2,左眼可見Schwalbe線。輔助檢查:NCT:OD 33mmHg,OS 43mmHg,給予鹽酸卡替洛爾滴眼液、布林佐胺滴眼液點(diǎn)眼后,眼壓:右眼12mmHg,左眼17mmHg。臨床診斷:右眼白內(nèi)障,左眼Peters綜合征,雙眼繼發(fā)性青光眼,雙耳聽力損害,室間隔修補(bǔ)術(shù)后。
2.2基因突變篩查 基因突變篩查結(jié)果顯示,該患者c.788G>A位點(diǎn)突變,而該基因的其他編碼序列及外顯子與內(nèi)含子的交界處均未發(fā)現(xiàn)序列異常。該突變位于第五外顯子內(nèi),編碼區(qū)的第788個(gè)堿基由G突變成A,直接導(dǎo)致了其編碼蛋白的改變,致使原蛋白的第69位氨基酸由精氨酸變成了組氨酸,正常及突變患者測(cè)序圖對(duì)照見圖3。該患兒家屬中的正常個(gè)體及無(wú)親緣關(guān)系的100位正常對(duì)照測(cè)序分析均未發(fā)現(xiàn)同樣基因的突變,文獻(xiàn)檢索未見PPS病基因突變位點(diǎn)的報(bào)道。
2.3 PITX2蛋白的生物信息學(xué)分析 本研究中我們?cè)?例PPS患者中發(fā)現(xiàn)一新的PITX2基因突變,該突變?yōu)殡s合突變,編碼區(qū)的第788個(gè)G堿基突變成A,對(duì)c.788G>A突變進(jìn)行分析顯示,該位點(diǎn)堿基的突變位于第五外顯子內(nèi),直接導(dǎo)致了其編碼蛋白的改變,致使原蛋白的第69位氨基酸由精氨酸變成了組氨酸。通過PolyPhen軟件對(duì)該錯(cuò)義突變導(dǎo)致氨基酸變化的可能致病性進(jìn)行分析,結(jié)果顯示該變與致病性之間具有一定的關(guān)聯(lián)性(軟件分析結(jié)果:benign,0.003)。進(jìn)一步用Phyre2軟件構(gòu)建c.788G>A突變前后的蛋白分子三級(jí)結(jié)構(gòu)模型(圖4)。
3討論
3.1 Peters綜合征臨床特點(diǎn) PPS是先天性角膜混濁眼病較為典型的類型,多見于嬰幼兒或青少年期,其主要表現(xiàn):先天性角膜中央部混濁白斑,伴有相應(yīng)區(qū)域的角膜后部基質(zhì)變薄和后彈力層缺損,虹膜角膜粘連,前房較淺,周邊部角膜厚度可正常,角膜緣后可見后胚胎環(huán),青光眼發(fā)病率在50%,小眼球伴發(fā)率達(dá)40%[1],另外可有眼部非特征性異常如前葡萄腫、鞏膜化角膜、虹膜缺損及原始玻璃體增生癥等表現(xiàn)。大多數(shù)是散發(fā)病例,但也有一些常染色體顯性及隱性遺傳的報(bào)道。該病通常被認(rèn)為是一種單獨(dú)的眼部疾病,但實(shí)質(zhì)上35%患者可伴發(fā)全身發(fā)育的異常[2],如并發(fā)率在15%左右的生長(zhǎng)發(fā)育遲緩、唇裂、心臟室間隔缺損、耳畸形、聽力損害、中樞神經(jīng)或泌尿生殖系統(tǒng)異常等。本研究中的該突變患者病例臨床特點(diǎn):(1)先天起病;(2)角膜中央變薄,白色混濁,后彈力層缺損;(3)虹膜角膜粘連,角膜緣后可見后胚胎環(huán);(4)繼發(fā)性青光眼;(5)同時(shí)伴有室間隔缺損以及聽力損害、全身發(fā)育的異常。以上臨床特點(diǎn)符合PPS的臨床診斷[3]。目前PPS引起的青光眼可以采用藥物或手術(shù)治療,小梁切除術(shù)輔助抗代謝藥物應(yīng)用是首選。如角膜混濁局限,可長(zhǎng)期散瞳治療。如角膜混濁致密,應(yīng)在出生后3mo內(nèi)行穿透性角膜移植術(shù),如果合并嚴(yán)重白內(nèi)障,可同時(shí)行白內(nèi)障手術(shù)以防止形覺剝奪性弱視。本例突變患兒,雙眼青光眼予以鹽酸卡替洛爾滴眼液、布林佐胺滴眼液點(diǎn)眼后眼壓控制良好,患兒家屬考慮予以藥物保守治療;右眼白內(nèi)障則行白內(nèi)障超聲乳化及人工晶狀體植入術(shù),左眼Peters綜合征引起的角膜混濁行穿透性角膜移植術(shù),目前觀察患兒雙眼術(shù)后恢復(fù)良好。
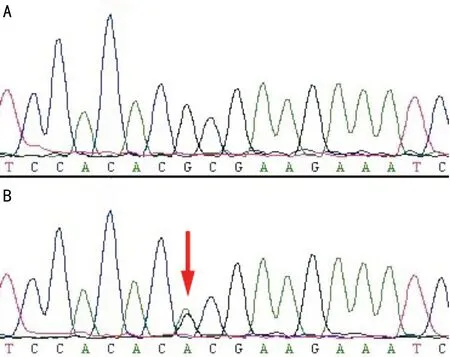
圖3 正常及突變患者測(cè)序圖 A:正常;B:突變。
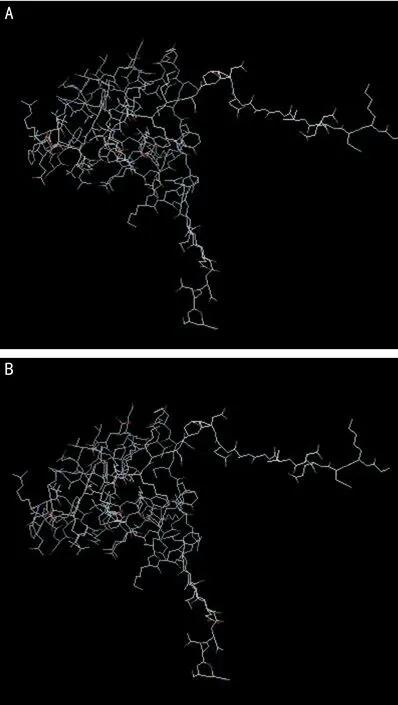
圖4 PITX2野生型及突變型蛋白質(zhì)分子三級(jí)結(jié)構(gòu)模型 A:野生型;B:突變型。
3.2 PITX2基因功能和突變與Peters綜合征發(fā)病機(jī)制 PITX2屬于Paired-Bicoied同型盒蛋白家族,并與PITX1和PITX3構(gòu)成PTX亞家族,定位于4q25-q27,全長(zhǎng)19 930bp,形成271aa、317aa和324aa三種蛋白質(zhì)亞型。這三種亞型主要區(qū)別在于氨基端的不同,而同源結(jié)構(gòu)域以及羧基端的序列則完全相同。其包含一個(gè)60氨基酸同源結(jié)構(gòu)域2(homeobox-2)和一個(gè)14氨基酸構(gòu)成OAR(otp aristaless and rax)結(jié)構(gòu)域,前者可通過其內(nèi)在的“螺旋-旋轉(zhuǎn)-螺旋”(helix-turn-helix,HTH)結(jié)構(gòu)與特異性的DNA位點(diǎn)相結(jié)合,在個(gè)體發(fā)育過程中間發(fā)揮著重要作用,該同源結(jié)構(gòu)域第50位氨基酸恒定為賴氨酸,對(duì)于特異性結(jié)合DNA具有決定性。后者推測(cè)與轉(zhuǎn)錄激活、蛋白質(zhì)結(jié)合DNA有關(guān)。Peters綜合征國(guó)外報(bào)道在新生兒中的發(fā)病率在1.2/100 000,致病基因定位在11P13區(qū)域PAX6基因和4q25區(qū)域PITX2基因,目前國(guó)外僅有較少Peter異常由PITX2基因突變致病的報(bào)道。1999年英國(guó)的Doward首次報(bào)道PITX2基因突變導(dǎo)致Peters綜合征后,日本報(bào)道了1例Peters綜合征患者的PITX2的突變[4];在國(guó)內(nèi),Peters綜合征未見任何PITX2基因突變的報(bào)道。在本研究中,我們對(duì)10例先天性角膜混濁的患者進(jìn)行PITX2基因突變的篩查,1例患者檢測(cè)到一新的基因突變c.788G>A,臨床特點(diǎn)及PITX2基因突變的研究均符合PPS的臨床診斷, 這是中國(guó)首次報(bào)道PPS PITX2的基因突變。該堿基突變位于Exon5內(nèi),編碼區(qū)的第788個(gè)堿基由G突變成A,該位點(diǎn)堿基的突變直接導(dǎo)致了其編碼蛋白的改變,致使原蛋白的第69位氨基酸由精氨酸變成了組氨酸。推測(cè)PITX2基因的突變是PITX2蛋白結(jié)合不同靶基因DNA的關(guān)鍵區(qū)域,因此該位置突變將引起兩者結(jié)合的不穩(wěn)定性,從而影響蛋白質(zhì)的功能。
3.3 Peters綜合征基因型與表型的研究 PPS與Axenfeld-Rieger綜合征(簡(jiǎn)稱ARS)是兩種不同類型的眼前段發(fā)育不良疾病,最新的研究證實(shí)胚胎學(xué)形成機(jī)制均是由神經(jīng)嵴細(xì)胞的間充質(zhì)細(xì)胞遷移分化異常引起。轉(zhuǎn)錄因子PITX2及FOXC1是這兩種眼病的共同致病基因,基因的突變使神經(jīng)嵴細(xì)胞的遷移分化過程出現(xiàn)異常,則會(huì)對(duì)眼前節(jié)的組織結(jié)構(gòu)產(chǎn)生廣泛的影響,從而導(dǎo)致PPS和ARS等眼前段疾病的發(fā)生[5];神經(jīng)嵴細(xì)胞在心臟、骨、顱軟骨、牙、真皮的形成發(fā)育中如受到阻礙,便可引起患者室間隔缺損以及聽力損害等全身異常。國(guó)外文獻(xiàn)曾報(bào)道了同一家系中FOXC1同一突變位點(diǎn)突變引起PPS和ARS兩種眼前段發(fā)育不良的臨床表型[6];有意思的是,國(guó)外文獻(xiàn)報(bào)道了PITX2基因的同一位點(diǎn)c.788G>A突變導(dǎo)致Axenfeld-Rieger綜合征病例[7],而本研究中的測(cè)序結(jié)果發(fā)現(xiàn)的同樣位點(diǎn)c.788G>A突變導(dǎo)致PPS臨床表型,PITX2可引起兩種不同眼前段發(fā)育不良ARS和PPS眼病,似乎意味著兩者眼病發(fā)病原因間的共同聯(lián)系。在眼前段胚胎學(xué)形成發(fā)育過程中,多種基因的信號(hào)轉(zhuǎn)導(dǎo)通路相互聯(lián)系,形成復(fù)雜的網(wǎng)絡(luò)體系,一個(gè)基因功能的實(shí)現(xiàn)依賴于該基因與其它基因間的相互作用,這些基因通過相互協(xié)作來控制生物體重要的生命過程,由此我們推測(cè)兩種不同眼病的發(fā)生是由等位基因的突變或是PITX2和傳導(dǎo)通路中的其他基因如FOXC1在共同的信號(hào)傳導(dǎo)中的相互影響而導(dǎo)致。
PPS是臨床少見病,但患者可伴發(fā)極高的青光眼發(fā)病幾率,是嚴(yán)重影響患者視力的致盲性眼病,而國(guó)內(nèi)資料較少,國(guó)內(nèi)眼科醫(yī)師對(duì)PPS認(rèn)識(shí)不足,臨床中經(jīng)常被誤診。本研究分析了Peters綜合征的臨床特點(diǎn),對(duì)其致病基因PITX2進(jìn)行突變檢測(cè),闡明其發(fā)病的分子遺傳學(xué)病因,以了解其基因型與表型的關(guān)系。今后需要更大規(guī)模、更深入地研究Peters綜合征的病因,以及病因與臨床表型之間的關(guān)系,從而為該眼病的診斷預(yù)防和治療起到更全面的指導(dǎo)作用。
1 Mayer UM. Peters Anomaly and Combination with Other Malformations-(Series of 16 Patients).OphthalmicPaediatrGenet1992;13(2):131-135
2 Heon E, Barsoumhomsy M, Cevrette L,etal. Peters Anomaly - the Spectrum of Associated Ocular and Systemic Malformations.OphthalmicPaediatrGenet1992;13(2):137-143
3 Ozeki H, Shirai S, Nozaki M,etal. Ocular and Systemic Features of Peters’ Anomaly.GraefesArchClinExpOphthalmol2000;238(10):833-839
4 Arikawa A, Yoshida S, Yoshikawa H,etal. Case of Novel Pitx2 Gene Mutation Associated with Peters’ Anomaly and Persistent Hyperplastic Primary Vitreous.Eye2010;24(2):391-393
5 Sowden JC. Molecular and Developmental Mechanisms of Anterior Segment Dysgenesis.Eye2007;21(10):1310-1318
6 Weisschuh N, Wolf C, Wissinger B,etal. A novel mutation in the FOXC1 gene in a family with Axenfeld-Rieger syndrome and Peters’anomaly.ClinGenet2008;74(5):476-480
7 Kulak SC, Kozlowski K, Semina EV,etal. Mutation in the Rieg1 Gene in Patients with Iridogoniodysgenesis Syndrome.HumMolGenet1998;7(7):1113-1117
Clinical and genetic research in Chinese families with Peters syndrome
Li-Qin Huang1, Guo-Hua Lu1, Yang Xie1, Ping-An Mao1, Xin-Cheng Sun1, Yong Meng2
Foundation item:Guidance Project of Changzhou Municipal Health Bureau (No.WZ201315)1Department of Ophthalmology, Changzhou No.2 People’s Hospital, Changzhou 213000, Jiangsu Province, China;2Department of Ophthalmology, Changzhou No.3 People’s Hospital, Changzhou 213000, Jiangsu Province, China
Yong Meng. Department of Ophthalmology, Changzhou No.3 people’s Hospital,Changzhou 213000, Jiangsu Province, China. 13861047902@163.com
?AIM: To research the clinical characteristics and identify the disease-causing gene mutation in Chinese patients with Peters syndrome. All these cases will be useful foundations for clinical diagnosis, medical therapy and pathogenesis.
Peters syndrome; congenital corneal opacities; PITX2; mutation
常州市衛(wèi)生局指導(dǎo)性項(xiàng)目(No.WZ201315)
1(213000)中國(guó)江蘇省常州市第二人民醫(yī)院眼科;2(213000)中國(guó)江蘇省常州市第三人民醫(yī)院眼科
黃麗琴,主治醫(yī)師,研究方向:眼遺傳病。
孟永,主治醫(yī)師,研究方向:眼底病.13861047902@163.com
2016-08-03
2016-11-08
:Huang LQ, Lu GH, Xie Y,etal. Clinical and genetic research in Chinese families with Peters syndrome.GuojiYankeZazhi(IntEyeSci) 2016;16(12):2237-2240
10.3980/j.issn.1672-5123.2016.12.16
Received:2016-08-03 Accepted:2016-11-08
猜你喜歡
英語(yǔ)世界(2023年6期)2023-06-30 06:29:10
中國(guó)醫(yī)學(xué)影像學(xué)雜志(2021年6期)2021-08-13 08:43:36
中國(guó)生殖健康(2020年2期)2021-01-18 02:51:26
小學(xué)生導(dǎo)刊(2018年13期)2018-06-29 03:49:00
中國(guó)生殖健康(2018年2期)2018-01-12 13:57:51
現(xiàn)代檢驗(yàn)醫(yī)學(xué)雜志(2016年4期)2016-11-15 02:01:14
中國(guó)現(xiàn)代醫(yī)學(xué)雜志(2015年26期)2015-12-23 11:04:22
鄭州大學(xué)學(xué)報(bào)(醫(yī)學(xué)版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國(guó)神經(jīng)精神疾病雜志(2014年1期)2014-03-01 03:23:22
