分子系統發育分析的生物信息學方法
2016-12-23 06:47:12郭一冉
山西農經 2016年12期
□郭一冉
(襄陽市第五中學湖北襄陽441057)
分子系統發育分析的生物信息學方法
□郭一冉
(襄陽市第五中學湖北襄陽441057)
處于信息時代,計算機技術滲入到生物學研究中,研究人員開始從分子水平對生物進化進行研究,并運用計算的方法針對為生命起源的研究探索出新的思路。多年來,從分子的層面對物種的系統發育進行研究取得了一定的成果。本論文針對分子系統發育分析的生物信息學方法進行研究。
物種進化;分子系統發育;計算機技術;生物信息學
生命的進化是漫長的。史學界針對物種的進化史進行研究,都試圖從生物化石中尋找物種進化的證據,但畢竟化石數量有限,而且通過這種方式所獲得的進化信息是零散的。所以,要對生物的進化史以及生物之間的進化關系充分掌握,目前的學術界會普遍采用解剖學、發育學的相關理論進行研究。但是,這些研究方法都不同程度地存在著依賴性,這就必然會導致研究中存在著局限性。生物的結構相似,但是,進化的途徑并不完全相同。比如,魚類和脊椎動物的眼睛所發揮的功能是相同的,但是,進化的途徑卻是完全不同的。隨著分子生物學的發展,對物種進化的研究就可以從生物分子的層面展開,以獲得更為準確的物種進化信息。
1 物種進化研究中分子系統發育分析所發揮的作用
系統發育學又被稱為“系統發生學”,主要的研究內容是物種形成的歷史和進化的歷史,而且還針對物種在進化過程中相互之間所存在的關系進行研究。在生物信息學研究領域中,系統發育學是重要的分支。在對物種進化進行研究的過程中,從系統發育學的角度進行研究,可以對物種的進化史更好地掌握,基于此而對生命的起源進行探索,包括物種的變異、物種的差異、物種的基因功能以及從生態學的角度對微生物的研究等等。
隨著生物學的研究進入到分子層面,基因技術開始融入到生物進化史研究中。特別是基因測序技術的發展,諸如RNA、DNA以及蛋白質等等的生物序列逐漸積累起來,這就使得生物進化史研究進入到分析層面。在很多生物學專家看來,在生物分子中就可以獲得物種進化的信息,而且相比較于從生物化石獲取信息要容易得多[1]。所以,生物研究領域對于物種的進化進行研究,多會從分析層面展開。
隨著學界對物種發育的研究采用生物信息學的方法,能夠涉及到的研究學科越來越多,除了計算機技術和生物學之外,包括數學、統計學等等都被用于研究中,從分子的層面對生物進化史研究水平逐漸提高,而且在研究方法上不斷實現創新。
2 分子層面的物種進化信息
2.1 單條生物序列中所含有的進化信息
如果生物的基因或者蛋白質均為同源的,當從一條序列向另一條序列進化的時候,對于進化的概率進行計算,就需要通過變異的次數對物種進化的距離進行衡量。刻畫單條序列的分子進化的過程中所產生的信息,就是計算局部位點上所存在的堿基變異情況或者是氨基酸殘基上所存在的變異情況,所有的進化事件,包括進化信息的插入、進化信息的刪除以及進化信息的轉化等等,都會詳細記錄下來。
在提取進化信息的時候,從單基因水平進行提取,就是將能夠對物種進化情況有所反映的基因提取出來,通過比較不同物種之間的基因而獲得兩條基因序列所存在的不同之處。不同物種的基因序列差異越小,就意味著物種之間所存在的進化距離就越近。
2.2 多條生物序列中所含有的進化信息
對于多條生物序列中所含有的進化信息進行研究,主要采用兩種方法。其一,在系統發育樹的構建上采用單序列信息,用于表示物種系統;其二,采用比對的算法從多條生物序列的角度對同源基因進行比對,之后串聯所獲得的結果。根據所獲得的比對結果將系統發育樹進行重新構建[2]。同源基因被找出來之后,就將這些基因信息充分利用起來,并對這些信息進行分類。
比如,對神經嵴細胞采用生物信息學的方法對基因差異進行分析,可以利用DAVID數據庫對與基因有關的數據進行富集,并根據需要予以分類。DAVID數據庫可以對500個基因所發生的改變情況進行生物信息學分析,具體操作:打開DAVID網頁進入到指定的數據庫中,將發生改變的神經嵴細胞基因提取出來,從原有的表格中復制到具有統計功能的基因輸入框中。數據提交完畢后,選擇“Start Analysis”并點擊,就可以對這500個基因進行生物信息學分析了。(下圖:神經嵴細胞分化)
3 采用系統發育樹針對物種進化關系進行研究
3.1 建立在字符序列基礎上而采用的系統發育樹算法
建立在字符序列基礎上而采用的系統發育樹算法是將可以發揮各種功能的樹搜索出來,選擇對給定序列能夠給予很好的解釋的樹,用以對物種的系統發育進行研究。
3.1.1 最大簡約法。最大簡約法以通過最小的改變對物種群體之間所存在的差異進行觀察。在對發育樹的選擇上,要選擇進化次數最小的那棵樹而對物種進化關系進行研究。多年來,采用這種方式對生物的進化情況進行研究,隨著物種數量的增多,這種方法由于沒有對樹中的分支進行掌握,導致物種進化的距離無法明確地反映出來。
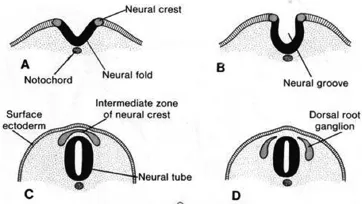
神經嵴細胞分化
3.1.2 最大似然法。最大似然法所采用的是進化模型,通過將模式數據與真實的數據信息之間對比,統計相似程度。最大似然法的數據統計效果良好,其不僅對物種進化的距離充分考慮,還對距離的相關內容進行了刻畫。但是,采用這種方法需要對發育樹分支的拓撲結構進行研究,計算過程非常復雜。如果物種的數量大,采用這種方法很顯然是不適宜的。
3.1.3 貝葉斯推斷法。貝葉斯推斷法是基于最大后驗概率原理,通過所掌握的先驗知識對后驗的分布情況進行求解。要求所選擇的發育樹為最大后驗概率,對發育樹為真的概率進行分析,并采用貝葉斯法進行推斷。這種方法被廣泛地應用。但是,在推斷的過程中,需要對先驗概率進行估計,還要對各種參數進行集成,所以,在計算的時候需要消耗大量的時間,所以,貝葉斯推斷法存在著局限性。
3.2 基于物種進化距離的系統發育樹算法
基于物種進化距離的系統發育樹算法中,較為經典的是兩種算法,即,UPJMA法和鄰接法。其中的鄰接法屬于是合并算法,雖然這種算法并不能將計算結果精確到最小進化樹,但是可以獲得近似的數值,不僅計算的速度快,而且具有較高的準確率。基于物種進化距離而采用鄰接法,可以使得計算的過程和所獲得的結果更容易被理解,與常規的字符序列方法相比,不僅計算的速度上存在著優勢,而且還可以將物種距離的矩陣計算出來,之后就能夠采用聚類算法將物種的發育樹構建起來。
結束語
隨著信息技術的發展,計算機技術逐漸滲入到生物進化史研究中。計算機具有很強的數據處理能力,在對生物進化相關的數據進行處理的時候,不僅數據處理能力提高了,而且數據處理成本有所降低。所以,采用生物信息學方法對分析系統發育系統進行分析非常必要。
[1]詹永勤,余敏,楊長平.關于中美生物信息學研究現狀的研究[J].西南農業學報,2013(02):789—794.
[2]盧境婷,王旭東,代杰文,等.顱神經嵴細胞的遷移及特性[J].中華口腔醫學研究雜志,2011,5(06):58—61.
1004-7026(2016)12-0103-02
Q75
A
10.16675/j.cnki.cn14-1065/f.2016.12.078
猜你喜歡
英語世界(2023年10期)2023-11-17 09:18:18
天天愛科學(2022年9期)2022-09-15 01:12:54
天天愛科學(2022年4期)2022-05-23 12:41:48
當代水產(2022年3期)2022-04-26 14:26:56
航空世界(2020年10期)2020-01-19 14:36:20
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
汽車觀察(2018年10期)2018-11-06 07:05:26
中華手工(2017年2期)2017-06-06 23:00:31
少兒科學周刊·少年版(2015年1期)2015-07-07 17:15:12
中外會展(2014年4期)2014-11-27 07:46:46
