基于物化性質對嗜熱蛋白的預測
2017-04-10 06:13:27刀福英陳欣欣
生物信息學 2017年1期
關鍵詞:特征
刀福英,陳欣欣,林 昊
(神經信息教育部重點實驗室信息生物學中心(電子科技大學生命科學與技術學院),成都610054)
基于物化性質對嗜熱蛋白的預測
刀福英,陳欣欣,林 昊*
(神經信息教育部重點實驗室信息生物學中心(電子科技大學生命科學與技術學院),成都610054)
嗜熱蛋白在高溫下能保持穩定性和活性,是研究蛋白質熱穩定性的理想模型,開發一個蛋白質熱穩定性識別的方法將對蛋白質工程和蛋白質的設計很有幫助。目前的研究中,氨基酸的組成及其物化性質一直被認為和蛋白質的熱穩定性相關。本研究篩選出可靠的數據集,包括915個嗜熱蛋白和793個非嗜熱蛋白。利用蛋白質氨基酸的物化性質和氨基酸的組成表征嗜熱蛋白,將二肽氨基酸組成整合到9組氨基酸物化性質中使蛋白序列公式化。支持向量機5折疊交叉驗證表明:當gap=0時,290個特征產生的精度最高,為92.74%。因此說明對于分析蛋白質的熱穩定性,所建立的預測模型將是一個很有效的工具。
嗜熱蛋白;熱穩定性;偽氨基酸組分;氨基酸物化性質
嗜熱和嗜冷微生物是兩種重要的極端微生物, 存在于其中的嗜熱和嗜冷酶是基礎研究和工業應用的熱點,它有助于認知蛋白質折疊、蛋白質結構和功能的關系以及設計用于極端環境的生物催化劑。隨著第一個極端嗜熱微生物Methanococcusjannaschii基因組的公布, 研究者通過比較基因組(蛋白質組)的方法對其穩定性機制進行了深入的探討。近年來, 不少嗜冷微生物的基因組測序工作陸續完成, 使得對嗜熱和嗜冷蛋白穩定性機理的研究不斷深入。盡管研究者對上述極端蛋白穩定性機理的探討較多,但利用蛋白質序列信息對其嗜熱和嗜冷特性的理論預測卻很少。
從蛋白質序列出發對其高級結構及特性進行理論預測所面臨的一個重要課題是如何有效提取蛋白質序列特征, 氨基酸組成是最常用的一種方法, 此外, 利用二肽組成和偽氨基酸組成在一些情況下也取得了較好效果。在后基因組時代,隨著DNA和蛋白質序列及結構信息的大量積累,人們利用數學、計算機科學的知識分析、挖掘生物數據,以尋求蘊涵在其中的生物學規律。
基于蛋白質序列特性可以對嗜熱蛋白進行預測,Liang等[1]使用氨基酸耦合模型去區分嗜熱與嗜常溫蛋白,Zhang等[2]利用二肽和氨基酸組分來區分嗜熱與嗜常溫蛋白,其中五折交叉驗證精度達86.6%,后來Gromiha和Suresh[3]將他們的數據去除冗余后,在神經網絡的基礎上運用氨基酸組分得到的五折交叉驗證精度達到了89%。Montanucci等[4]運用支持向量機去預測蛋白質熱穩定性,jackknife交叉檢驗的預測精度為88%。Wu等[5]提議運用決策樹來預測蛋白質熱穩定性,其預測精度在80%以上。盡管以上這些研究都獲得了好的結果,但預測精度還有待提高。
在本文的研究中,構建了包括915個嗜熱蛋白和793個非嗜熱蛋白在內的很可靠的標準數據集,運用氨基酸二肽組分和九組氨基酸物化性質來表征蛋白質的特征,通過方差分析來進行特征篩選,利用支持向量機區分嗜熱與非嗜熱蛋白。本文使用的特征篩選技術可以提高預測精度,經過優化的290個參數的五折疊交叉驗證準確率達到了92.74%,Jackknife交叉驗證結果顯示有91.69%的嗜熱蛋白和91.42%的非嗜熱蛋白是正確預測的,其ROC曲線面積為0.963。因此表明本文構建了較為精準的模型,可以通過對未知蛋白的序列預測其耐熱性,從而可以判斷其是否具有熱穩定性,是否可以運用于相應的酶工程之中。
1 材料與方法
1.1 構建數據集
在當前的研究中,嗜熱蛋白和非嗜熱蛋白分別從嗜熱有機體和非嗜熱有機體中提取的。為了保證當溫度上升到嗜熱生物的溫度時使獲得的非嗜熱蛋白變性,將60 ℃作為嗜熱有機體最適生長溫度的最低溫度限制,將30 ℃作為非嗜熱有機體最適生長溫度的最高溫度限制,對NCBI里1 126個全微生物基因組生物的最適生長溫度進行篩選,有136個原核基因組(包括17個古生菌和119個細菌)滿足要求。
從UniProt中根據最適溫度的標記分別從136個原核有機體中選取嗜熱和非嗜熱蛋白序列,為了保證得到數據的可靠性則需要滿足以下篩選步驟:(1)蛋白質必須是經過手動注釋和審核的;(2)排除蛋白質序列中具有歧義的殘基(例如帶有“X”,“B”和“Z”);(3)排除含有其他蛋白片段的序列;(4)排除從預測或同源關系中推論的缺少可信度的蛋白質。嚴格遵照以上4個程序篩選得到1 329個嗜熱蛋白和1 250個非嗜熱蛋白。
這里構建的初步數據集中,通常還會存在一些冗余序列。數據集如果由許多相似度較高的樣本組成,那么較高的冗余度就會導致統計代表性降低。如果預測器由一個有偏倚的數據集訓練而來,更有可能產生錯誤的高估結果。為了除去冗余并避免偏倚,使用了CD-HIT軟件[6]來篩選這些序列片段。
CD-HIT的基本思路是先對所有數據集里的序列,根據序列的長度從長到短進行排序,以最長的一條序列作為第一個序列類。然后依次處理排好序的各條序列,CD-HIT不僅能夠對單獨的數據集執行去除冗余信息,還可以比較兩個不同的數據集。
本文選取的一致性閾值為0.4,去除序列相似度性在40%以上的序列后,最終的數據集包括915個嗜熱蛋白和793個非嗜熱蛋白,最終獲得了1 708個樣本作為基準數據集S,用公式表示如下:
S=ST∪Snon-T
(1)
這里的兩個子集分別包含915個嗜熱蛋白樣本和793個非嗜熱蛋白樣本,符號“∪”表示兩個子集的并集。
1.2 特征提取
在嗜熱蛋白的預測中,用有效的數學公式來規定蛋白質序列是一個很有效的方法。一個很直接的辦法是將公式表示全部蛋白質序列的全部氨基酸,公式如下:
P=R1R2R3…RL
(2)
其中R1、R2、R3......RL分別表示蛋白質樣本P中的第1個,第2個,第3個,…… ,第L個氨基酸殘基,有了這樣的公式,就可以被很多序列相似搜索工具用來進行數據的預測,比如BLAST、FASTA等,對于一個高的相似序列的數據集,它的預測結果往往是很好的,所以這樣的基于相似的方法是很直觀的,但是有一個不可忽視的問題,在訓練的數據集中查詢序列的相似序列如果不能被找到的話它是就不會起作用。因此在對蛋白分類時提議利用離散向量表示蛋白質樣本。偽氨基酸組分表示蛋白質序列是一個被廣泛使用的方法,偽氨基酸組分(PseACC)[7]是一種能夠很好地表征蛋白質序列的信息參數。它不但能夠描述蛋白質序列的氨基酸組成,而且能夠描述蛋白質氨基酸序列的物理化學性質的關聯。基于偽氨基酸組分的概念,本文做了一個提升,將二肽氨基酸組分代替氨基酸組分,并且進行十組這樣的特征提取,即gap值從0取到9,表示兩氨基酸殘基間間隔從0到9。
將g-gap二肽氨基酸組成來代替氨基酸組成,所以此參數不僅反映了兩類蛋白在序列的組成和序列順序的區別外,還能表現出殘基間相關性,在基準數據集中將 400+nλ維向量表示每個蛋白質,表示公式如下:
P=[x1…x400x400+1…x400+nλ]T
(3)
其中
(4)
(5)
在公式(4)中,fu表示蛋白質P中二肽氨基酸的標準頻率,公式(5)中nu表示蛋白質P中第u個二肽氨基酸的數量,很容易理解的二肽的數量總共有400(20×20)個,用二肽氨基酸頻率來表征蛋白質的特征。
下面對氨基酸物化性質進行描述。公式(4)τu中的表示序列物化性質的相關性,由以下公式計算得到:
(6)
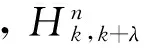
(7)
(8)
1.3 特征篩選
由公式(3)可知,用400+nλ個特征表示每個蛋白,為了能夠得到最優的特征集,使用方差分析來進行特征篩選,將特征值進行排序,由以下公式來對特征打分:
(9)
在該公式中xu(i,j)表示在第i類樣本中第j個樣本的第u個特征的頻率值;mi表示第i類樣本的樣本數(本文共有兩類樣本,m1=915為嗜熱蛋白,m2=793為非嗜熱蛋白)。分析該公式可知第u個特征對應的打分值F越大表明該特征區分嗜熱蛋白與非嗜熱蛋白的能力越強,因此將F值作為特征篩選標準。
1.4 支持向量機
根據耐熱性對蛋白質進行預測就是蛋白質分類的過程。分類的方法很多,如費歇爾判別式,神經網絡,集成學習,k-鄰近算法等被廣泛用于蛋白質的分類中。對小樣本的分類本文使用支持向量機來構建分類器。
支持向量機(Support Vector Machine,SVM)[8]是目前極其流行的數據挖掘的工具。SVM的基本思想有如下兩個方面:首先,支持向量機對線性條件下可以進行分類情況進行分析研究。當遇到線性條件下無法進行分類時,理論上應該把訓練樣本通過某種非線性的映射對數據進行升維處理,這樣就會把數據升為較高維度的特征向量空間,在此空間中,尋找出線性的最佳超平面;其次,支持向量機的思想是建立在結構風險最小化的理論之上,支持向量機需要在高維空間中尋找分類超平面,尋找兩種類別的樣本點之間的最大分類間隔。本文通過網格搜索進行5折疊交叉驗證,找到最佳的特征。支持向量機可以由libsvm軟件包來運行。
1.5 評估指標
在統計學預測檢驗中,對于一個給定的基準數據集,jackknife檢驗[9]能夠產生獨一無二的結果,所以在實際應用中它經常被用來評估方法的性能。為了節省計算時間,本文在特征篩選的過程中使用5折疊交叉檢驗,挑選出最佳的特征集之后運用jackknife檢驗再次對特征集計算檢驗。它可在敏感性(Sn),特異性(Sp),準確率(Acc),馬修相關系數(MCC)4個方面來評估。這4個參數由以下公式計算得到:
(10)
(11)
(12)
MCC=
(13)
Sn,Sp,Acc的范圍為[0,1],MCC范圍為[-1,1]。這里FN(False Negative)表示被判定為負樣本,但事實上是正樣本;FP(False Positive)表示被判定為正樣本,但事實上是負樣本;TN(True Negative)表示被判定為負樣本,事實上也是負樣本;TP(True Positive)表示被判定為正樣本,事實上也是正樣本。(本文正樣本為嗜熱蛋白,負樣本為非嗜熱蛋白)。這4個指標通常被用在統計預測理論中,它們可以從4個不同的角度來定量的衡量預測系統的性能。
此外,受試者特征曲線(ROC曲線)能兼顧靈敏度和特異性要求以綜合評價分類器的預測性能,曲線下面積作為量化指標可以直觀有效的比較不同分類器的性能優劣。線上的每個點都是對同一個分類器預測的反應,通常由于不同的判斷標準得出了一系列不同的預測結果。受試者操作特征曲線的橫坐標軸通常為虛報概率,縱坐標軸一般為擊中概率,根據測試數據在特定分類器的不同的判斷標準下得到的不同結果繪制出曲線。
2 結果與討論
2.1 物化性質
在蛋白質的結構和功能中氨基酸的物化性質扮演著十分重要的角色,氨基酸的六種物化性質被廣泛使用,分別是氨基酸的疏水性、親水性、氨基酸側鏈基團質量、-COOH基團的解離常數、-NH3基團解離常數、25℃時的等電點,在本文的研究中,除了以上六種物化性質外,還添加三種氨基酸物化性質,分別是氨基酸的剛性、柔性、不可替代性。九組氨基酸的物化性質[10]運用于公式(4)~(8)中。
在蛋白質的結構和功能中,氨基酸側鏈基團的硬度和靈活性包含著重要有用的信息,剛性與柔性值是通過主成分分析獲得的[11]。在生物的進化中,有些殘基是很容易被替代的,但有些殘基卻很難被替代,不可替代性可由氨基酸的平均突變危險性來描述[12],平均突變危險性值越高表示該殘基越難以被替代,不可替代性反應了在生命進化過程中的突變危險性。
2.2 預測精度
基于上面介紹的九組物化性質,本文可以得到400+9λ個特征,即在公式(3)~(6)中n=9,為了能夠包含盡可能多的相關信息,節省計算資源,本文取λ=10,因此,用490維向量表示每個數據集中的每個蛋白質樣本。
為得到最好的預測性能,挑選出具有最大精度的最佳特征,如果研究所有的特征,就會得到一個最好的特征集,但是490個特征的所有可能的組合的數目太大了,超出了大部分計算機的計算能力,所以要做到檢驗所有特征組合的性能那是不可能的,為節省計算時間,運用公式(9)中F打分來進行特征篩選,首先根據每個特征對應的F值從大到小進行排序,然后將第一個特征即具有最大F值的特征用SVM計算其精度,接下來,按照F值從大到小對應的特征值依次加到前一個特征集,依次每次進行SVM計算該特征集的精度,這個過程要一直重復,直到最小F值特征包含到該特征集中,即一共包含490個特征。所以最后SVM計算會產生相應的490個精度,分別是按照F值從大到小排列后的第一個特征對應的精度,前兩個特征對應的精度,前三個特征對應的精度直到得到490個特征對應的精度為止,比較得到的精度,會得到一個最高精度對應的特征集。基于特征篩選技術,高維數據將會投射到低維空間,本文用該最佳的特征集來構建最終的預測模型。
變化參數gap的值分別取0到9,所以需要計算4 900(490×10)個特征集對應的精度,將特征數作為橫坐標,將精度作為縱坐標,在笛卡爾坐標系中得到10組曲線圖。如圖1所示,當gap=0,橫坐標290特征對應的精度為92.74%,該精度為最高精度。用jackknife檢驗計算該包含290個特征的模型,得Sn=91.69%,Sp=91.42%,表明該模型能夠正確識別嗜熱蛋白。
為了用這290個特征一目了然的描繪該模型的性能,在圖2中繪制了ROC曲線,從圖中可以看出曲線靠近左邊和頂部坐標軸,表明該模型適用于嗜熱蛋白與非嗜熱蛋白的分類,在jackknife交叉檢驗中ROC曲線下的面積值為0.963。
為了對比,基于相同的數據集,還通過WEKA用了樸素貝葉斯[13]、貝葉斯網絡、隨機森林[14-15]三種方法進一步計算分類性能,預測結果顯示在表1中,比較表1中的數據,很明顯可以看出SVM是預測嗜熱蛋白的最好的算法。
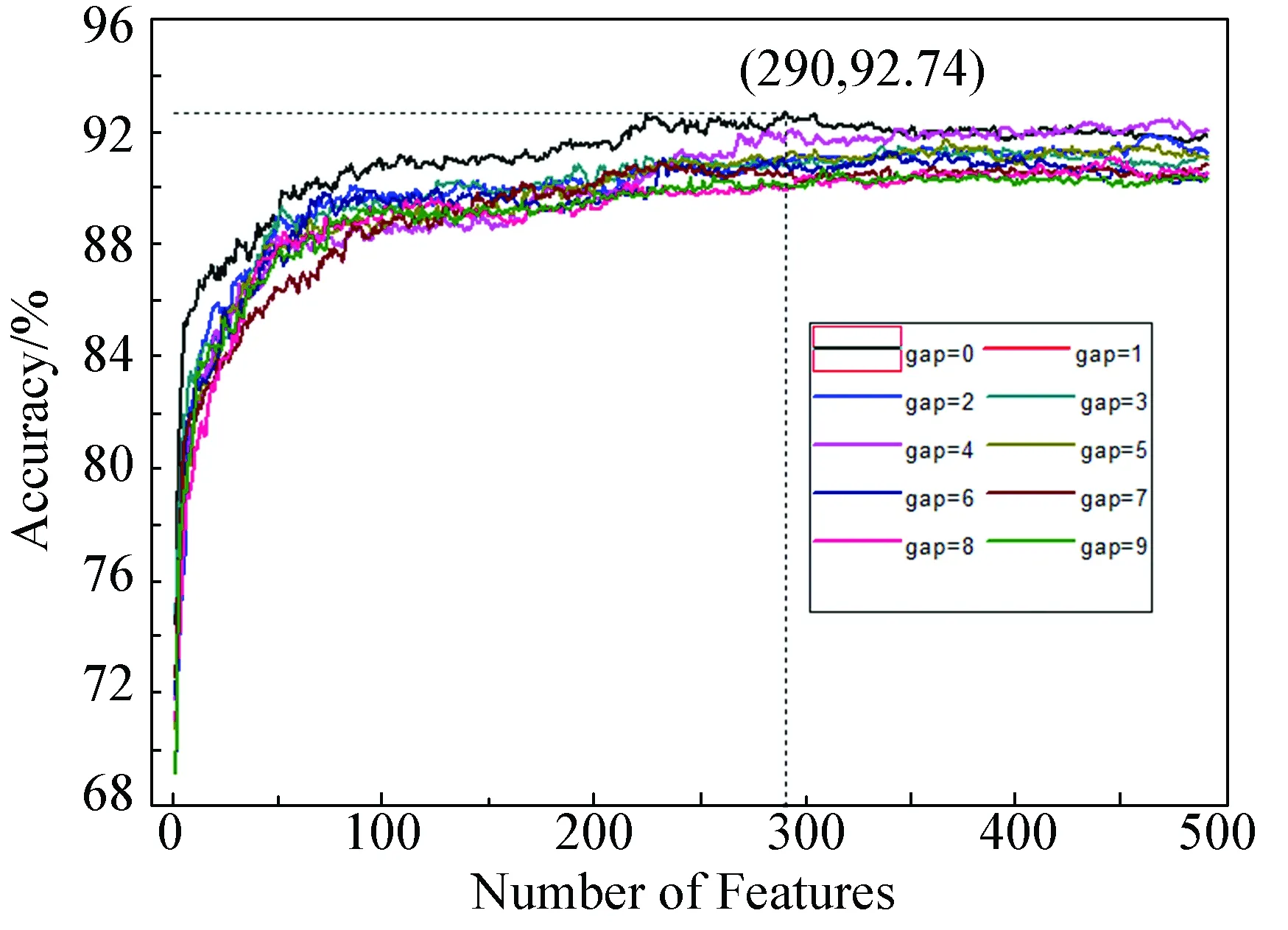
圖1 特征篩選結果*Fig.1 A plot to show the feature selection results
*彩圖見電子版(http://swxxx.alljournals.cn/ch/index.aspx)(2017年第1期DOI:10.3969/j.issn.1672-5565.2017.01.201606001)
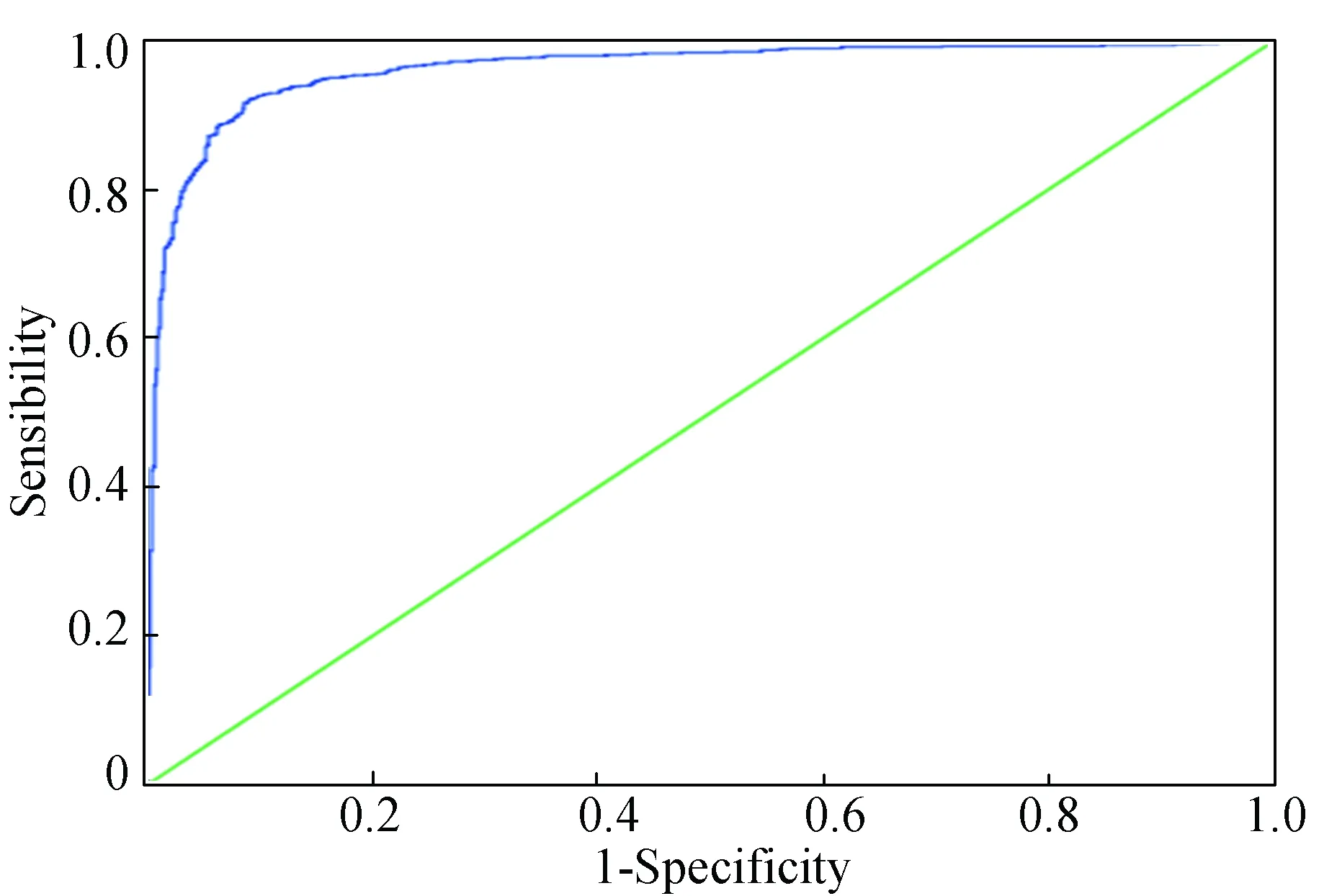
圖2 最佳的290個特征在jackknife交叉驗證中的ROC曲線Fig.2 The ROC curve for the model with 290 optimal 0-gap dipeptides in the jackknife cross-validation
注:對角線表示ROC的面積為0.5。

表1 比較不同算法的結果Table 1 Comparing the performance of different algorithms
3 總結與展望
蛋白質的熱穩定性與酶工程密切相關,對蛋白質熱穩定性的研究將對蛋白質工程和蛋白質的設計很有幫助,因此,開發了一個從非嗜熱蛋白中篩選識別出嗜熱蛋白的方法,獲得了高精度的模型。結果表明,該方法可以篩選有效的特征,提高預測性能,在優化模型的基礎上,將建立一個在線的預測網絡服務器,便于識別嗜熱蛋白。在嗜熱蛋白分析和進一步的實驗研究中,這個預測將成為一個很有用的工具,此外,在這項研究中提出的方法可以推廣到其他蛋白質的預測中。
為了能夠得到更高精度的預測模型,接下來需要從以下方面來進行工作:
(1)實時更新搜索數據集,完善擴大數據集。比如可以將數據集篩選標準的最適溫度范圍擴大。
(2)提取新特征。例如還可以提取氨基酸、三肽、四肽甚至多肽作為特征,或者選取不同的物化性質作為特征等,篩選后尋求最佳精度,提高預測模型精度。
(3)開發更加準確、快速的分類預測算法。比如可以將隨機森林和支持向量機相結合等。
(4)拓展研究。可以將蛋白質的熱穩定性理論與其他生物學過程相結合進行研究,例如可以研究蛋白質的亞細胞定位與其耐熱性的關系、嗜熱菌在生物催化中的應用等相關領域。
References)
[1]LIANG H K, HUANG C M, KO M T, et al. Amino acid coupling patterns in thermophilic proteins[J]. Proteins,2005,59 (1): 58-63. DOI: 10.1002/prot.20386.
[2]ZHANG G Y, FANG B S. Application of amino acid distribution along the sequence for discriminating mesophilic and thermophilic proteins[J]. Process Biochemistry,2006,41(8): 1792-1798. DOI: 10.1016/j.procbio.2006.03.026.
[3]GROMIHA M M, SURESH M X. Discrimination of mesophilic and thermophilic proteins using machine learning algorithms[J]. Proteins,2008, 70(4): 1274-1279. DOI: 10.1002/prot.21616.
[4]MONTANUCCI L, FARISELLI P, MARTELLI P L, et al. Predicting protein thermostability changes from sequence upon multiple mutations[J]. Bioinformatics,2008,24(13): 190-195. DOI:10.1093/bioinformatics/btn166.
[5]WU L C, LEE J X, HUANG H D, et al. An expert system to predict protein thermostability using decision tree[J]. Expert Systems with Applications,2009, 36(5):9007-9014.DOI: 10.1016/j.eswa.2008.12.020.
[6]LI W Z, GODZIK A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences[J]. Bioinformatics, 2006,22(13):1658-1659. DOI: 10.1093/bioinformatics/btl158.
[7]CHOU K C. Prediction of protein cellular attributes using pseudo-amino acid composition[J]. Proteins, 2001,43(3):246-255.DOI: 10.1002/prot.1035.
[8]BHASIN M, RAGHAVA G P. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST[J]. Nucleic Acids Research,2004, 32(Web Server issue): W414-W419. DOI: 10.1093/nar/gkh350.
[9]CHOU K C. Some remarks on protein attribute prediction and pseudo amino acid composition[J]. Journal of Theoretical Biology, 2011,273(1): 236-247. DOI: 10.1016/j.jtbi.2010.12.024.
[10]TANG H, CHEN W,LIN H. Identification of immunoglobulins using Chou’s pseudo amino acid composition with feature selection technique[J]. Molecular BioSystems, 2016,12(4):1269-275.DOI:10.1039/C5MB00883B.
[11]GOTTFRIES J, ERIKSSON L. Extensions to amino acid description[J]. Molecular Diversity ,2010, 14(4):709-718. DOI: 10.1007/s11030-009-9204-2.
[12]LUO L F. The degeneracy rule of genetic code[J]. Origins of Life and Evolution of Biospheres,1988,18(1-2): 65-70. DOI:10.1007/BF01808781.
[13]丁彥蕊,蔡宇杰,孫俊,等.基于SVM 和KNN 的蛋白質耐熱性分類[J] 計算機工程與應用,2007,43(16):228-237.
DING Yanrui,CAI Yujjie,SUN Jun,et al. Protein heat tolerance classification based on SVM and KNN[J]. Computer Engineering and Applications,2007, 43(16):228-237.
[14]賈富倉,李華.基于隨機森林的多譜磁共振圖像分割[J]. 計算機工程, 2005,31(10): 159-161.
JIA Fucang,LI Hua. Multi spectral magnetic resonance image segmentation based on random forest[J]. Computer Engineering, 2005,31(10): 159-161.
[15]張光亞,方柏山. 基于氨基酸組成分布的嗜熱和嗜冷蛋白隨機森林分類模型[J].生物工程學報, 2008,24(2):302-308.
ZHANG Guangya, FANG Baishan. Based on the distribution of the amino acid composition is addicted to heat and psychrophilic protein random forest classification model[J]. Chinese Journal of Biotechnology,2008,24(2):302-308.
Prediction of thermophilic proteins based on physicochemical properties
DAO Fuying,CHEN Xinxin,LIN Hao*
(KeyLaboratoryforNeuro-InformationofMinistryofEducation,CenterforInformationalBiology,SchoolofLifeScienceandTechnology,UniversityofElectronicScienceandTechnologyofChina,Chengdu610054,China)
Thermophilic proteins can keep stability and activity at high temperature, which are ideal materials to study stability of proteins. Developing a valuable method to identify thermostability of protein would be helpful for protein engineering. In the present study, amino acid composition and physicochemical properties of protein have been thought of being related to the thermostability of protein. A reliable benchmark dataset including 915 thermophilic proteins and 793 non-thermophilic proteins is constructed for training and testing the proposed model in this article. We define protein samples using physicochemical properties and component of amino acid, so we design a descriptor which will combine dipeptide composition with nine physiochemical properties of amino acids. The results by support vector machine (SVM) with 5-fold cross-validation show that the best accuracy is 92.74% by using 290 features when the parameter gap is 0, indicating that our model holds very high potential to become a useful tool for the research on protein thermostability.
Thermophilic proteins;Thermostability;Pseudo amino acid composition;Physico-chemical roperties
2016-06-26;
2016-07-20.
四川省應用基礎研究項目(2015JY0100);中央高校基本業務費(ZYGX2015J144,ZYGX2015Z006)。
刀福英,女,碩士研究生,研究方向:生物信息學;E-mail:18200234053@163.com.
*通信作者:林昊,男,研究員,碩士生導師,研究方向:生物信息學;E-mail:hlin@uestc.edu.cn.
10.3969/j.issn.1672-5565.2017.01.201606001
Q51
A
1672-5565(2017)01-001-06
猜你喜歡
數學小靈通·3-4年級(2024年2期)2024-05-15 02:02:28
中學生數理化(高中版.高考數學)(2022年3期)2022-04-26 14:04:16
數學年刊A輯(中文版)(2020年1期)2020-05-19 00:30:36
空間科學學報(2020年2期)2020-04-01 03:50:40
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
中等數學(2019年8期)2019-11-25 01:38:14
當代陜西(2019年10期)2019-06-03 10:12:04
新聞傳播(2018年11期)2018-08-29 08:15:24
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
