基于微量DNA樣本的簡化甲基化文庫構建方法研究
2017-04-10 06:10:57魏冬凱徐康萍
生物信息學 2017年1期
關鍵詞:方法
魏冬凱,南 蓬,徐康萍,裘 鋒,3*
(1. 蘇州貝斯派生物科技有限公司 江蘇 蘇州 215123;2. 復旦大學生命科學學院 上海 200438;3.上海生物信息技術研究中心 上海 201203)
基于微量DNA樣本的簡化甲基化文庫構建方法研究
魏冬凱1,2,南 蓬2,徐康萍1,裘 鋒1,3*
(1. 蘇州貝斯派生物科技有限公司 江蘇 蘇州 215123;2. 復旦大學生命科學學院 上海 200438;3.上海生物信息技術研究中心 上海 201203)
RRBS(簡化甲基化測序)是一種有效研究DNA甲基化狀態的測序方案。文庫構建是該方案中最關鍵的實驗步驟之一,RRBS文庫構建往往因為酶切產物少,文庫構建步驟繁多等因素,導致DNA起始量需要1 ug以上。然而很多來源于人的樣本,如冰凍組織穿刺、石蠟切片、顯微微切割等,都往往只能獲得100 ng以下的DNA,無法滿足常規RRBS文庫構建的起始DNA總量要求,從而大大限制RRBS技術的應用范圍。本研究主要探討并設計了基于微量DNA樣本(<100 ng)的RRBS文庫構建策略,主要通過優化MspI酶切條件、DNA片段大小篩選、縮減反應步驟及減少DNA轉移次數等技術手段,大大提高了DNA回收率,進而建立了2種有效使用微量DNA樣品的RRBS文庫構建方案(即EA-Method和WB-Method方案)。EA-Method方案在處理100 ng左右的DNA時,有經濟、快速、高效的特點,但文庫質量略差于WB-Method方案;WB-Method方案可將DNA起始量降低至10 ng,并且文庫質量高。為驗證WB-Method方法的可靠性,研究人員選取了3種志愿者樣本(全血和口拭子),采用WB-Method同時進行常量和微量RRBS文庫構建,并進行Illumina Hiseq平臺測序,數據通過生物信息分析得到微量RRBS檢測的CpG,CHG,CHH中C堿基的甲基化比例與常量RRBS結果一致。同時,該方法可以大大降低DNA損耗及損傷,因而該方法可將RRBS技術應用到更廣泛的樣本類型中去,有效拓展RRBS的研究領域。
二代測序;RRBS;簡化甲基化;EA-M;WB-M
表觀遺傳學是指在不改變DNA 序列的條件下所發生的可遺傳基因表達的變化[1].表觀遺傳學主要包括DNA 甲基化、組蛋白修飾和染色質結構.脊椎動物中,DNA 甲基化表現為在DNA 甲基化轉移酶(DNA methyltransferase,DNMT)作用下,甲基基團合成到5′-CpG-3′中胞嘧啶的第五位碳原子上[2].正常情況下,人類基因組“垃圾”序列的CpG二核苷酸相對稀少,并且總是處于甲基化狀態。非甲基化的CpG不是均勻分布,而是呈現局部聚集傾向,形成一些GC含量較高、CpG雙核苷酸相對聚集的區域,即CpG島[3].與之相反,人類基因組中大小為100~1000 bp 左右且富含CpG二核苷酸的CpG島則總是處于未甲基化狀態,并且與56%的人類基因組編碼基因相關[4]。在正常人的基因組DNA中,約有3%~6%的胞嘧啶是甲基化的[5]. 根據Gardiner-Garden 等[6]的定義,CpG 島是一段長度不小于200 bp、GC 含量不小于50%、CpG 含量與期望含量之比不小于0.6 的區域.由于該定義將一些重復片段也包含其中,Takai 和Jones[7]將CpG 島重新定義為長度不小于500 bp、GC 含量不小于55%、CpG 含量與期望含量之比不小于0.65的區域.據統計,多于50%的基因的啟動子區含有CpG 島[8-9].在早期的認識中,CpG 島都是非甲基化的,但是隨著研究的不斷深入,人們發現在印跡基因、失活的X 染色體[10]甚至是正常的體細胞中都存在甲基化的CpG 島[11].部分CpG 島的異常甲基化常常伴隨著癌癥等疾病的發生[12]。高通量測序技術堪稱測序技術發展歷程的一個里程碑,該技術可以對數百萬個DNA 分子同時進行測序。這使得對一個物種的轉錄組和基因組進行全景式分析成為可能,因此也稱其為深度測序(Deep sequencing)[13]或下一代測序技術(Next generation sequencing,NGS)[14]。新一代高通量測序技術使得基因組整體水平高精度的甲基化檢測成為現實。目前,高通量測序技術已經廣泛應用于擬南芥、人、水稻和家蠶等生物DNA甲基化的研究,并取得了廣泛的研究成果[15-18]。
研究DNA 甲基化的高通量測序技術方法主要包括:全基因組重亞硫酸鹽測序(Bisulfite sequencing,Bi-seq) 和簡化表達重亞硫酸鹽測序 (Reduced Representation Bisulfite Sequencing, RRBS)、甲基化DNA 免疫共沉淀測序(Methylated DNA immuno precipitation sequencing, MeDIP-seq)、甲基化DNA 富集結合高通量測序(Methylated DNA binding domain sequencing, MBD-Seq)等。目前,重亞硫酸鹽修飾被公認為DNA 甲基化檢測的“金標準”。Bi-seq和RRBS技術均是基于上述修飾。全基因組Bisulfite 測序技術可以得到單堿基分辨率的全基因組甲基化圖譜。這一技術最初被Cokus等[15]應用在植物擬南芥的全基因甲基化圖譜分析上(借助了Solexa 測序技術)。2009年Lister 等[19]利用全基因組Bisulfite 測序發表了第一篇人類的全基因組甲基化圖譜( 利用了Solexa 測序),隨后該技術開始在人類疾病,如肥胖癥的研究中得以應用[20]。基于對黃種人的外周血單核細胞DNA 樣品進行Bisulfite 處理后,結合Solexa 測序技術進行深度測序,2010年Li等[21]成功繪制出了“炎黃一號”高精確度的全基因組甲基化圖譜。然而,重亞硫酸鹽轉化全基因組法高昂的測序成本,在一定程度上限制了該技術的廣泛應用。
簡化表達亞硫酸鹽測序(RRBS),又稱基于酶切消化的重亞硫酸鹽測序, 是一種簡便、高效、經濟的DNA甲基化研究方法,通過酶切富集啟動子及CpG島區域,并進行Bisulfite測序,同時實現DNA甲基化狀態檢測的高分辨率和測序數據的高利用率。2005年Meissner 等[22]最初結合Sanger 測序發明了該項技術,并將其成功應用于鼠胚胎干細胞去甲基轉移酶前后的甲基化譜的檢測。接著Meissner領導的研究小組通過改用MspI 酶切來富集小鼠全基因組近90%的CpG島片段,結合Illumina 公司的高通量測序技術,建立并完善了適用于哺乳動物全基因組甲基化測序分析的方法[23]。2010年該課題組又發表了一篇關于RRBS的文章,在其先前的工作基礎上進一步優化RRBS 技術并挖掘其在臨床應用上的潛力[24]。2012年華大基因研究院與深圳大學、中山大學及華南科技大學合作發表了RRBS與Bisulfite 測序的比較方法學文章[25]。Wang等[26]發現雙酶切(MspI,ApeKI)可以提高RRBS建庫成功率,并且提高覆蓋率。
目前RRBS構建測序文庫時所需DNA出發量一般大于100 ng[27],某些樣本甚至需要到達1 μg以上[28-29]。然而很多情況下實驗樣本很珍貴,如FFPE樣本或者單細胞樣本,這樣對RRBS建庫的要求和難度都大幅度提高,目前對于少于100 ng的樣本進行RRBS建庫缺乏有效的技術方法,本實驗對從微量DNA樣品出發構建RRBS文庫進行了較為細致的研究,并試圖找出并建立一種更簡便高效的方法。這對于拓寬簡化甲基化測序的應用領域有著重大意義。
1 材料與方法
1.1 材料
本實驗所使用的gDNA來源為實驗室自傳代人類紅白血病細胞系K562細胞,細胞采用懸浮培養至密度80%(K562細胞作為一種常見的研究用細胞系,具有一定代表性,并為保證細胞狀態最佳,所以將細胞培養至密度80%左右。),培養皿大小100 mm,培養基選用DMEM+10%FBS(胎牛血清),為保證gDNA來源保持一致,本實驗所用DNA采用一次性提取3個培養皿的混合K562細胞所得。
本實驗中,用于驗證WB-Method可靠性的人源樣本來源于3位志愿者,常量樣本采用靜脈血200 μL,微量樣本采用少量口腔上皮細胞(為驗證WB-M方案應用至真實人源樣本的可靠性,所以驗證實驗采用志愿者自采集樣本。)。
1.2 gDNA提取
gDNA提取方法采用蛋白酶K消化法,將細胞收集至15 mL離心管中,加入3 mL PBS緩沖液重懸細胞后,依次加入500 μL蛋白酶K(20 mg/mL)、7 mL 裂解液(SDS、EDTA、Tris-HCl),放置56 ℃孵育4 h后,1∶1(V/V)加入酚氯仿異戊醇(25∶24∶1)溶液,充分混勻后離心取上清,再按1∶1加入氯仿異戊醇(24∶1)溶液,充分混勻后離心取上清,加入0.8倍體積異丙醇(-20 ℃預冷)充分混勻后,可見白色團狀沉淀,使用槍頭將沉淀撈出,并使用70%乙醇漂洗沉淀2次,干燥揮發乙醇3 min后,使用Qiagen EB Buffer溶解沉淀。提取的DNA采用Qubit,NanoDrop和電泳進行質檢。
靜脈血gDNA提取方法使用QiaAmp Blood Mini Kit,按照操作手冊進行DNA提取,大致方法如下:將200 μL靜脈血置于冰水混合物中融化后加入20 μL蛋白酶K和600 μL AL緩沖液,混勻后置于56 ℃水浴15~30 min,裂解產物加入1倍體積乙醇充分混勻后過離心柱,所有產物過離心柱后依次加入AW1、AW2緩沖液洗滌離心柱2次,空氣干燥2~3 min,加入AE緩沖液洗脫DNA,提取的DNA采用Qubit,NanoDrop和電泳進行質檢。
1.3 MspI酶切條件優化
本實驗使用MspI限制性內切酶(NEB,20 U/μL)對gDNA進行酶切處理(因MspI酶切位點CCGG與人類CpG Island序列相似,并且該酶對甲基化區域不敏感,可以最大限度的富集CpG Island區域。),得到有效的簡化末端。因MspI酶對溫度不敏感,所以工作中只對MspI酶的用量和酶切時間進行條件優化實驗。優化實驗材料使用8 μg gDNA均分為7份,保證每份gDNA>1 μg,分別對每份gDNA,在37 ℃下采用不同時間和不同酶用量進行酶切,酶切體積50 μL,酶切完成后,65 ℃變性20 min,取25 μL酶切產物加入6X Loading Dye 5 μL,點2% 瓊脂糖凝膠 100 V 電泳2 h檢測酶切效率并回收相應片段。
1.4 片段大小篩選方法優化
片段大小篩選采用2種方法進行,分別是瓊脂糖凝膠電泳(加入Carrier DNA)(本實驗中所用的Carrier DNA為完全未甲基化的大腸桿菌DNA,unmethylation DNA,Promega)和磁珠兩步法篩選。
瓊脂糖凝膠電泳(加入Carrier DNA)方案,采用1 μg gDNA MspI 酶切后并進行補平和連接測序接頭所得的連接產物為材料,Carrier DNA采用unmethylation DNA (Promega)covaris打斷至150~300 bp,在連接產物中加入200 ng,不加入Carrier DNA的做為對照,進行2%瓊脂糖凝膠電泳,100 V 2h檢測加入CarrierDNA的效果。
磁珠兩步法篩選大小方案,方案原理基于AMpureXP Beads對PEG(MW 6 000)的終濃度敏感,從而影響結合DNA片段大小。本實驗采用1 μg gDNA covaris打斷至100~700 bp作為材料,不同PEG濃度進行片段大小篩選,并采用2%瓊脂糖凝膠電泳和BioAnalyzer 2100(毛細管電泳)進行片段大小檢測。
1.5 常規RRBS建庫方法
常規RRBS建庫方法 (Conventional Method, CO-M)(見圖1(a)),采用1 μg,100 ng,50 ng,10 ng gDNA,加1 μL (20 U/μL)MspI(以下所有使用酶試劑及酶反應buffer,均使用NEB來源,除特殊標注外)酶切 37 ℃ 0.5 h,加入末端修復反應試劑(10× Ligase Buffer 10 μL, 10 nM dNTPs 4 μL, 10 mM ATP 2.5 μL, T4 DNA Polymerase(3 U/μL) 5 μL,T4 PNK(10 U/μL) 5 μL,Klenow Large Fragment(5 U/μL) 1 μL,并補水至100 μL,20 ℃ 孵育30 min,產物用QiaAmp MinElute PCR Kit(Qiagen)進行純化并洗脫至30 μL,加入末端加A反應試劑(10mmol/L dATP 5 μL,Klenow Buffer 5 μL,5 U/μL Klenow exo-3 μL),補水至50 μL,37 ℃孵育30 min,產物用QiaAmp MinElute PCR Kit(Qiagen)進行純化并洗脫至30 μL,加入測序接頭連接反應試劑(甲基化處理的測序接頭(Illumina)1 μL,T4 ligase (10 U/μL) 3 μL,10×T4 ligase buffer 5 μL),加水補充至50 μL,22 ℃孵育15 min,連接產物使用QiaAmp MinElute PCR Kit(Qiagen)進行純化并洗脫至75 μL,重亞硫酸鹽CT轉化采用Zymo Gold Methylation Kit (Zymo Research),操作根據試劑盒說明書并將產物純化至20 μL,加入6× loading dye (Life Technologies)4 μL,2%瓊脂糖凝膠電泳回收150~350 bp片段,膠回收采用QiaAmp MinElute Gel Extraction Kit(Qiagen),回收至20 μL。加入文庫擴增試劑(Illumina Primer Mix 5 μL,2× KAPA uracil+ PCR Ready Mix (KAPA Biosystem) 20 μL,反應程序95 ℃ 10 min,擴增循環(95 ℃ 30 s,62 ℃ 30 s,72 ℃ 30 s)(1 μg DNA起始采用 10個循環,100 ng DNA及以下采用15個循環),72 ℃ 4 min),得到的PCR產物采用QiaAmp MinElute PCR Kit(Qiagen)進行純化至20 μL,Qubit測定文庫濃度,BioAnalyzer 2100(Agilent)檢測文庫片段分布。
1.6 EA一步RRBS建庫方法
EA一步建庫方法(EA Method, EA-M)(見圖1(b)),采用100 ng,50 ng,10 ng gDNA,加MspI(以下所有使用酶試劑及酶反應buffer,均使用NEB來源,除特殊標注外)1 μL酶切 37 ℃ 0.5 h,加入末端修復反應試劑(10× Ligase Buffer 2.5 μL, 10 nmol/L dNTPs 1 μL, 10 mmol/L ATP 2.5 μL, T4 DNA Polymerase (3 U/μL)2.5 μL,T4 PNK(10 U/μL) 2.5 μL, Taq Polymerase(10 U/μL) 1 μL),補水至25 μL,12 ℃15 min,37 ℃ 15 min,72 ℃ 20 min,產物不純化,直接加入測序接頭連接反應試劑(甲基化處理后的測序接頭(Illumina)1 μL,T4 ligase 3 μL,T4 ligase buffer 1 μL,加水補充至35 μL),22 ℃孵育15 min,連接產物使用QiaAmp MinElute PCR Kit(Qiagen)進行純化并洗脫至75 μL,重亞硫酸鹽CT轉化采用Zymo Gold Methylation Kit(Zymo Research) ,操作根據試劑盒說明書并將產物純化至20 μL,加入6× loading dye(Life Technologies) 4 μL,2%瓊脂糖凝膠電泳回收150~350 bp片段,膠回收采用QiaAmp MinElute Gel Extraction Kit(Qiagen),回收至20 μL。加入文庫擴增試劑(Illumina Primer Mix 5 μL,2× KAPA uracil+ PCR Ready Mix (KAPA Biosystem) 20 μL,反應程序95 ℃ 10 min,擴增循環(95 ℃ 30 s,62 ℃ 30 s,72 ℃ 30 s)(1 μg DNA起始采用 10個循環,100 ng DNA及以下采用15個循環),72 ℃ 4 min),得到的PCR產物采用QiaAmp MinElute PCR Kit(Qiagen)進行純化至20 μL,Qubit測定文庫濃度,BioAnalyzer 2100(Agilent)檢測文庫片段分布。
1.7 磁珠包被RRBS建庫方法
磁珠包被RRBS建庫方法 (WB Method, WB-M)(見圖 1(c)),采用100 ng,50 ng,10 ng gDNA,加1 μL MspI酶(以下所有使用酶試劑及酶反應buffer,均使用NEB來源,除特殊標注外) 37 ℃ 酶切過夜,加入末端修復試劑(10× Ligase Buffer 10 μL, 10 nmol/L dNTPs 4 μL, 10 mmol/L ATP 2.5 μL, T4 DNA Polymerase(3 U/μL) 5 μL,T4 PNK(10 U/μL) 5 μL, Klenow Large Fragment(5 U/μL) 1 μL),補水至100 μL,20 ℃ 孵育30 min,產物用150 μL Ampure XP Beads(Beckman)純化,純化步驟按照AmpureXP Beads說明書進行至揮發晾干殘留乙醇,加EB Buffer(Qiagen)37 μL,室溫放置1 min,無需去除磁珠,直接加入末端加A反應試劑(10 mmol/L dATP 5 μL,Klenow Buffer 5 μL,5 U/μL Klenow exo-3 μL),37 ℃孵育30 min,產物加入PEG/NaCl(20% PEG8000(Sigma), 9% NaCl(Sigma))溶液 50 μL,重懸磁珠,并按照AmPureXP Beads說明書進行至揮發晾干殘留乙醇,加EB Buffer(Qiagen)41 μL,室溫放置1 min,無需去除磁珠,加入測序接頭連接反應試劑(甲基化處理后的測序接頭(Illumina)1 μL,T4 ligase(10 U/μL) 3 μL,T4 ligase buffer 5 μL),加水補充至50 μL,22 ℃孵育15 min,產物加入PEG/NaCl(20% PEG8000, 9% NaCl)溶液 50 μL,重懸磁珠,并按照AmPureXP Beads說明書進行至揮發晾干殘留乙醇,加EB Buffer(Qiagen)75 μL,室溫放置1 min,無需去除磁珠,重亞硫酸鹽CT轉化采用Zymo Gold Methylation Kit 并純化至20 μL(本步驟使用Zymo試劑盒自帶純化柱,將磁珠去除),產物采用AmpureXP Beads 二步篩選法篩選片段大小至150~350 bp之間,采用EB Buffer洗脫至20 μL,加入文庫擴增反應試劑(Illumina Primer Mix 5μL,2× KAPA uracil+ PCR Ready Mix (KAPA Biosystem) 20 μL,反應程序95 ℃ 10 min,擴增循環(95 ℃ 30 s,62 ℃ 30 s, 72 ℃ 30 s)(1μg DNA起始采用 10個循環,100 ng DNA及以下采用15個循環),72 ℃ 4 min),得到的PCR產物采用AmpureXP Beads進行純化至20 μL,Qubit測定文庫濃度,BioAnalyzer 2100檢測文庫片段分布。
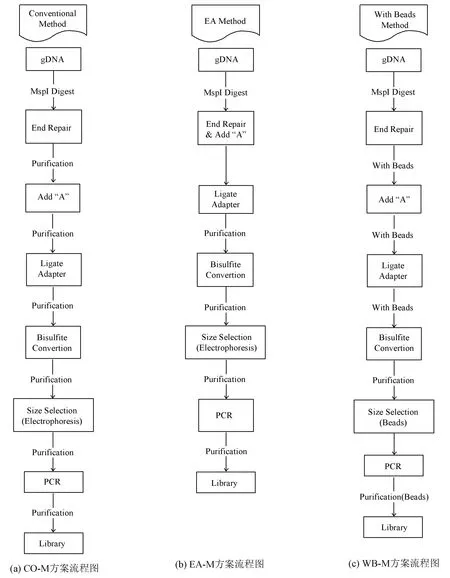
圖1 三種RRBS文庫構建方法流程圖Fig.1 Three methods for RRBS library construction
1.8 WB-Method驗證實驗
本實驗使用人來源樣本3例,采用WB-Method分別進行微量(10 ng)RRBS和常量(1 μg)RRBS文庫構建,并使用Illumina Hiseq 3000平臺進行高通量測序,每個樣本測序20 mol/L reads以上。生物信息分析使用Trim_galore進行低質量(Q<20)數據過濾和去重,bismark進行序列比對,參考基因組選用人類基因組hg19,并對甲基化頻率分布、甲基化覆蓋度、3種context(CpG、CHG、CHH)下C堿基甲基化比例進行統計。
2 結果
2.1 gDNA提取及質量測定
gDNA提取方法采用經典PK法(細胞和組織樣本),全血采用QiaAmp使用Blood Mini Kit,Qubit測定濃度并計算K562 gDNA總量為386 μg(Qubit 2.0 HSDNA Assay),Nanodrop 2000檢測gDNA純度(OD260/OD280=1.91,OD260/OD230=2.12 ),0.6% 瓊脂糖凝膠檢測gDNA純度,電泳條帶清晰單一,無降解拖尾條帶,片段均在40 kb以上(見圖2)。人來源的每個常量樣本均獲得2 μg以上DNA(OD260/OD280=1.94,OD260/OD230=2.08),微量樣本獲得100 ng以上DNA(OD260/OD280=1.90,OD260/OD230=2.16),純度均符合質檢標準(1.8
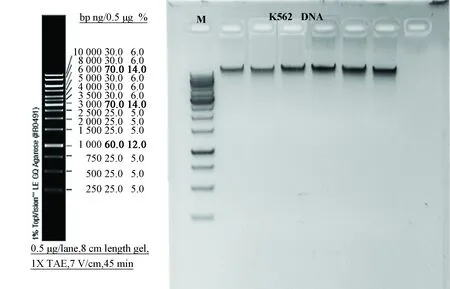
圖2 基因組DNA電泳質量檢測Fig.2 gDNA electrophoresis
2.2 MspI酶切條件優化
本實驗提及的3種RRBS文庫構建方法都需要使用MspI酶切富集CpG Island位點,所以需要優化MspI酶切條件,酶切條件選用37 ℃做為酶切溫度,選擇酶切時間1 h設置酶用量10 U,20 U,40 U,再根據MspI的酶切特性,選擇10 U的酶活設置酶切時間條件為0.5 h,1.0 h,2.0 h,4.0 h,O/N(16 h)。通過電泳圖(見圖3)可以看到,酶用量加大和酶切時間延長并不能讓MspI的酶切效率提高,所以從時間和成本的角度出發,酶切條件使用10 U,0.5 h,酶切溫度37 ℃,這里需要說明,10 U的酶切用量已經可以將1 μg DNA酶切完整,所以本課題所用100 ng DNA使用10 U,0.5 h,37 ℃的酶切條件完全可以消化完全。
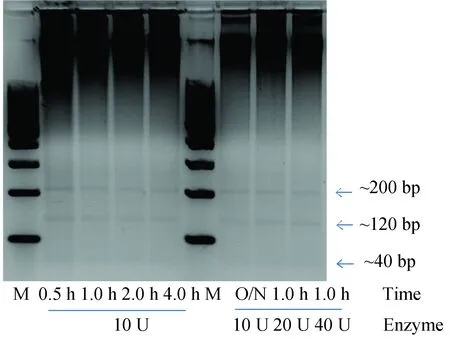
圖3 MspI酶切條件優化電泳檢測Fig.3 MspI digest condition optimization
2.3 片段大小篩選方法優化
二代測序儀在測序時需要進行成簇反應(Cluster Generarion),成簇反應對DNA片段長度有一定要求,根據Illumina官方解釋,正常情況下,成簇反應需求DNA片段長度在200~500 bp之間,所以文庫片段過大或過小都會影響成簇反應進行,這也使得篩選合適的DNA片段大小成為文庫構建中不可缺少的一步。另外,片段篩選的另一個作用是去除接頭二聚體,接頭二聚體對測序質量會造成極大影響,據統計,5%的測序接頭二聚體含量會導致最終數據50%的損失。片段篩選最常見的方法是瓊脂糖凝膠電泳,本課題因起始DNA量很少,導致連接反應的產物在瓊脂糖凝膠上基本不能顯現,導致切膠時容易誤判并且膠回收效率降低,所以本文采用了加入Carrier DNA的方式提高辨識度和膠回收效率,由于在片段大小篩選前,已經使用重亞硫酸鹽處理過連接產物,Carrier DNA因不含測序擴增接頭序列,在最終的PCR擴增中不會被擴增,所以不會影響最終的測序結果。本實驗采用加入Carrier DNA和未加入Carrier DNA進行對照實驗,主要比較在瓊脂糖凝膠(見圖 4(a))上的可辨識度,圖上可以很清楚的加入Carrier DNA的目的電泳片段區域(150~350 bp)較未加入Carrier DNA的目的電泳片段區域清晰,切膠時可以明顯的將接頭二聚體與目的序列分開,有效提高辨識度并且在膠回收過程中,提高了回收效率,且Carrier DNA可以填補硅膠柱中的死體積,從而達到提高回收效率的目的。
使用加入Carrier DNA的方法可以提高膠回收的得率,但微量DNA通過瓊脂糖凝膠后的損失率仍然可達到50%以上,為了進一步減少損失,采用了羧基磁珠(Ampure XP Beads)進行文庫大小篩選,篩選原理是因羧基磁珠在PEG的影響下,結合DNA的能力會產生變化,PEG含量越高,磁珠可結合的片段就越多,從片段大小上來看,能結合的小片段就越多,使用不同濃度的PEG對打斷后的DNA進行篩選,并使用瓊脂糖凝膠電泳檢測分布(見圖4(b)),由圖中可以看到,12%~15%的PEG濃度已經可以滿足RRBS文庫構建的需要,為驗證該方案的可重復性,繼續設計優化實驗如下:取1μg Fragment DNA(200~600 bp),分別用12%/15%篩選和10%/12%篩選,并采用BioAnalyzer進行片段大小分析,做1個重復(見圖4(c)),從2 100檢測圖種可以看到,12%/15%可用于RRBS文庫構建中的片段大小篩選,與瓊脂糖凝膠分離切膠方法相比,得率和穩定性有較大提高(見圖4(c))。
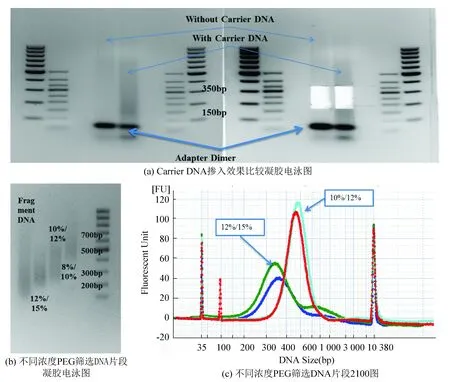
圖4 片段大小篩選方法優化Fig.4 Size Selection Optimization
2.4 EA Method 與 Conventional Method比較
EA Method(EA-M)方案主要是將CO-M中的末端修復及3’端加dA合并成一步,并且在做完末端修復和3’端加dA之后,直接加入T4 ligase,減少了其中2步純化步驟,從而減少DNA損失。該方法可以將末端修復及3’端加dA步驟合并使用了Taq Polymerase在72 ℃下可以在3’端加dA的特性,將Taq Polymerase加入到末端修復的體系中即可實現3’端加dA的目的,并且將這一步的buffer替換成T4 DNA Ligase Buffer,這就使得在進行下一步連接操作時,不需進行純化。本實驗使用100 ng,50 ng,10 ng gDNA,每個實驗組設置3個重復,共9個樣本,分別使用CO-M和EA-M進行RRBS文庫構建,本實驗中PCR控制12個循環數,并使用Qubit測定并統計比較2種方法的文庫總量(見圖5(a)) ,CO-M法在處理10 ng,50 ng gDNA時文庫構建失敗,在處理100 ng DNA時3個樣本的其中1個失敗,2個樣本得率很低,EA-M法在處理10 ng DNA時得率很低,文庫質量差,處理50 ng,100 ng DNA時已能得到很好的結果。
本實驗使用1 μg gDNA起始CO-M進行RRBS文庫構建作為文庫合格的陽性參照,該陽性參照使用Qubit測定文庫濃度并計算總量為151.2 ng,并使用BioAnalyzer 2100分別測定陽性文庫(見圖5(b))和EA-M構建的文庫(見圖5(c)),使用EA-M法構建的RRBS文庫與陽性對照文庫相比,大小均分布在200~550之間,沒有接頭二聚體(Adapter dimer),圖中看到BioAnalyzer2100(Agilent)檢測出的峰不呈現良好的正態分布,是因RRBS文庫構建中DNA片段化使用MspI酶切產生的酶切末端導致,不會對后續測序結果產生任何影響。
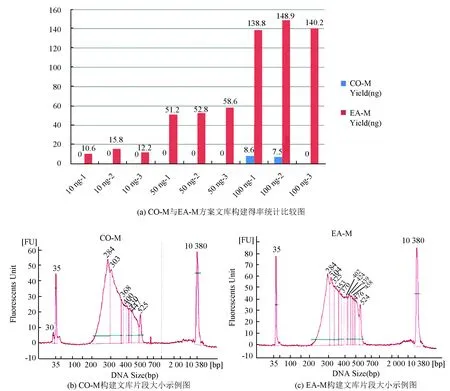
圖5 EA-M與CO-M構建RRBS文庫比較*Fig.5 EA-M vs CO-M
*注:(a)∶ EA-M和CO-M構建RRBS文庫,DNA起始分別使用10 ng,50 ng,100 ng,藍色*標注使用CO-M建庫,紅色*標注使用EA-M建庫;(b)∶ CO-M 文庫大小分布;(c)∶ EA-M文庫大小分布。
彩圖見電子版(http∶//swxxx.alljournals.cn.ch/login.aspx)(2017年第1期doi∶10.3969/j.issn.1672-5565.2017.01.201609001)。
2.5 EA Method穩定性測試
根據EA Method與Conventional Method比較試驗的結果,發現EA Method在處理50 ng DNA起始量時,已經有很好的效果。為了進一步測試EA Method的穩定性,將600 ng gDNA平均分成12份(每份50 ng),采用EA Method進行RRBS文庫構建,使用Qubit測定文庫得率并統計CV(見圖 6),發現EA Method在處理50 ng DNA進行RRBS文庫構建時,成功率高,文庫產出也很穩定(CV<0.05)。
2.6 WB Method與EA Method比較
由上述研究可以看出,EA-M法對于微量樣本的處理已初具優勢,但因為需要進行切膠和多次DNA轉移導致處理50 ng以下的樣本仍比較困難,為了能進一步降低DNA出發量和優化文庫質量,在磁珠篩選文庫大小方案的啟發下,建立了全程將DNA與磁珠包被的文庫構建方案,該方案的優勢在于,DNA全程與磁珠結合,大大減少DNA轉移的次數,提高最終有效文庫得率。該方案與CO-M法的步驟基本相同,但在所有的酶反應中,DNA都與磁珠結合在一起進行孵育,純化方案中,采用PEG Buffer進行結合和洗脫,在連接反應前,使用的PEG濃度均為20%,連接反應中采用12%/15%進行洗脫,本實驗采用了EA-M法作為對照,分別取10 ng,50 ng,100 ng做2組重復,RRBS文庫構建完成后,Qubit進行測定并統計比較2種方法的文庫得率(見圖7(a)),BioAnalyzer 2100分別隨機抽取2種方法中的1個文庫進行文庫片段大小測定(見圖7(b),7(c)),發現WB-M法在處理更微量DNA樣本上比EA有更大的改善,基本可以達到3倍左右的文庫總量,并且得到的文庫范圍與EA法有高度一致性(見圖7(a)~圖7(c))。
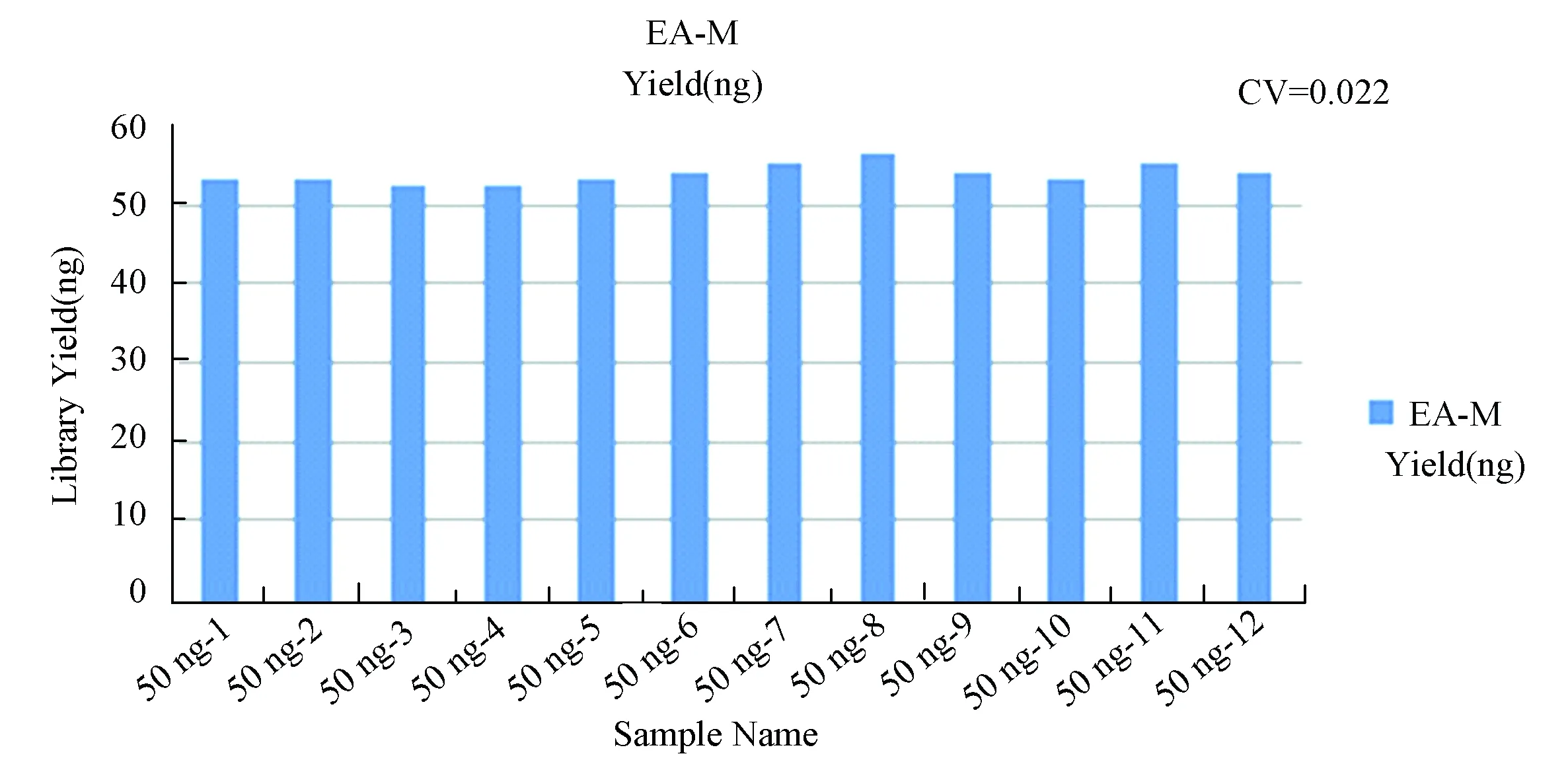
圖6 EA方案穩定性測試Fig.6 EA Method stability test
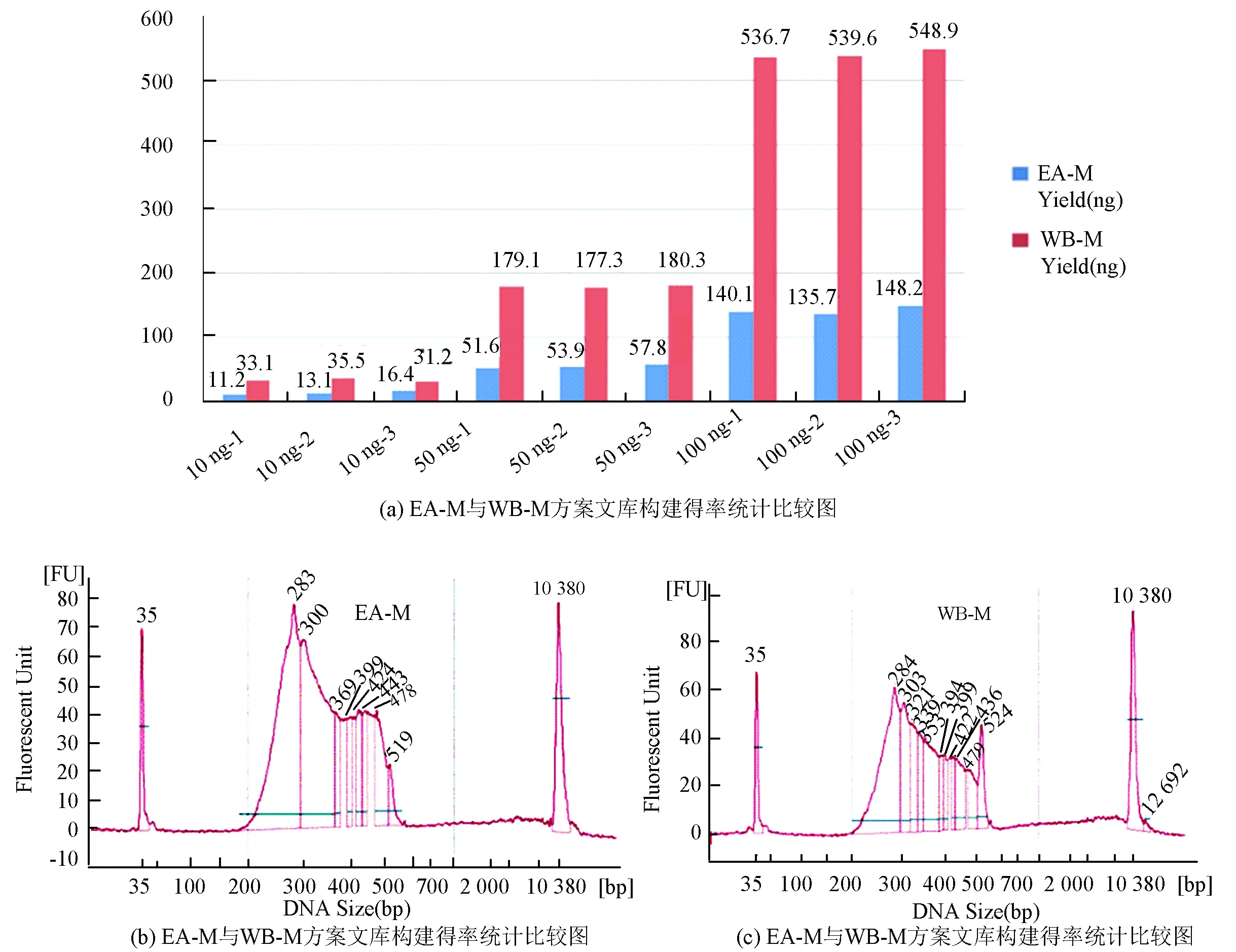
圖7 WB-M與EA-M構建RRBS文庫比較*Fig.7 WB-M vs EA-M
*注:(a)∶ EA-M和CO-M構建RRBS文庫,DNA起始分別使用10 ng,50 ng,100 ng,藍色*標注使用CO-M建庫,紅色*標注使用EA-M建庫;(b)∶ CO-M 文庫大小分布;(c)∶ EA-M文庫大小分布。
彩圖見電子版(http∶//swxxx.alljournals.cn.ch/login.aspx)(2017年第1期doi∶10.3969/j.issn.1672-5565.2017.01.201609001)。
2.7 WB Method穩定性測試
根據WB Method與EA Method比較試驗的結果,發現WB Method在處理10 ng gDNA起始量時,也有較為滿意的結果。為了進一步測試WB Method的穩定性,將120 ng gDNA均分成12份樣本(每份10 ng),采用WB Method進行RRBS文庫構建,使用Qubit測定文庫得率并統計CV,發現WB Method在處理10ng DNA進行RRBS文庫構建時,成功率高,文庫產出也很穩定(CV<0.05)(見圖 8)。
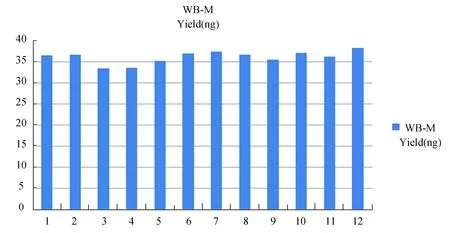
圖8 WB方案穩定性測試Fig.8 WB Method stability test
2.8 WB-M驗證實驗
使用WB-M對3個人來源樣本分別進行常量(1 μg)RRBS和微量(10 ng)RRBS文庫構建,并在Illumina Hiseq3000平臺上進行PE150測序,每個樣本至少獲得20 mol/L Reads數據。數據使用Trim_galore進行數據質量QC(見表1),可見3個常量樣本和3個對應微量樣本的QC結果一致,得到高質量(Q Score>20)數據后,再使用Bismark將數據比對至人類參考基因組(hg19版本),可看到常量與微量對應樣本數據Unique Reads均可達65%(見表1),再將得到的bam文件統計各個位點甲基化位點的甲基化比例及覆蓋度,并比較常量和微量樣本的差異,可見從不同甲基化水平堿基分布來看,微量和常量樣本均符合RRBS文庫的特點,甲基化區域均富集在Reads的兩頭(見圖9(c),9(d)),并且覆蓋CpG區域2X以上的reads占比80%以上(見圖9(a),圖9(b)),兩者保持了高度一致。最后統計了甲基化通常發生的CpG,CHG,CHH三種不同的context下C堿基的甲基化比例,其中CpG context最常見(見表2),可以看到常量和微量樣本的甲基化水平也保持一致。

表1 WB方案實驗數據QC Table 1 WB Method Data QC Analysis
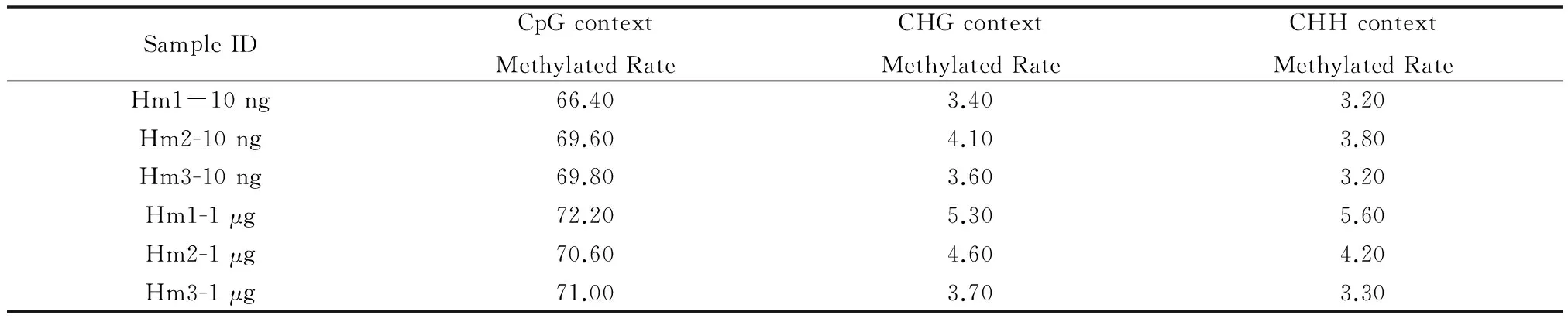
表2 WB方案CpG,CHG,CHH中C堿基甲基化比例Table 2 WB Method MethyC Content In CpG,CHG,CHH %
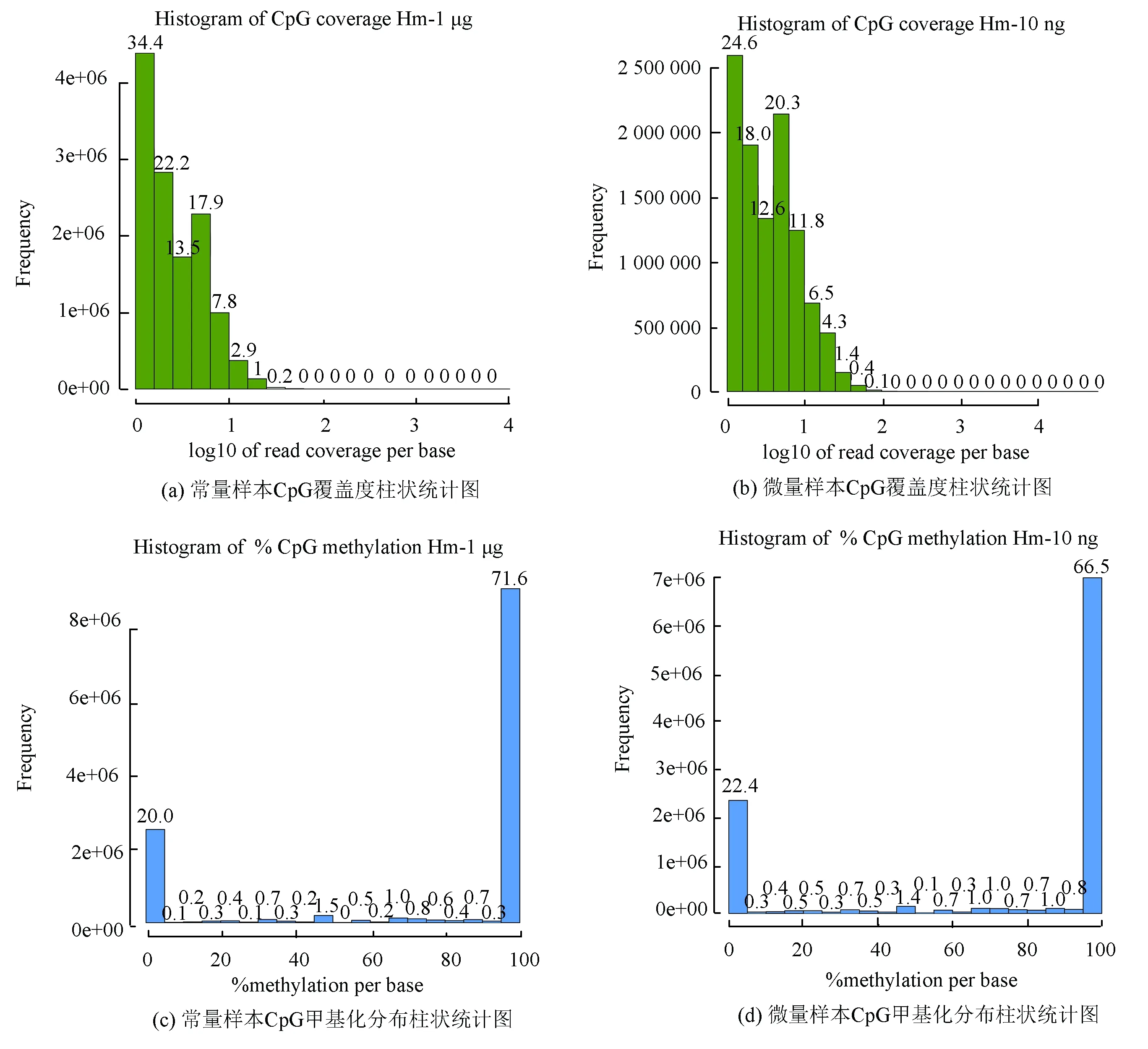
圖9 WB方案甲基化覆蓋度Fig.9 WB method methylation base coverage
3 討論
隨著表觀遺傳學研究的深入,二代測序在甲基化測序中的研究方法也大幅增多,RRBS(簡化甲基化)測序以經濟,分辨度高的優點在人、小鼠、大鼠的甲基化研究中被高頻使用。RRBS文庫是測序前樣本處理的關鍵步驟,其中的核心問題包括:(1)RRBS文庫需要符合MspI酶切的特性;(2)RRBS文庫必須沒有接頭二聚體存在(約125~128 bp);(3)文庫總量必須滿足二代測序基本需求(20 ng以上);(4)實驗方法的穩定性(可重復性)高。本研究中使用的2種改進后的基于微量DNA樣品的RRBS文庫構建方法,可以很好的解決以上4個問題,從而滿足RRBS文庫用于后續測序的需求。首先,經過MspI酶切優化實驗,確定了MspI酶的最優酶切條件,并且在最終文庫中也可以明顯的看到人gDNA被MspI 酶完整酶切的特征序列,并且文庫分布在200~600 bp,有利于在測序儀上進行成簇反應(cluster generation);其次,通過片段大小篩選優化方案,切膠純化通過加入carrierDNA 提高膠的辨識度,從而將接頭二聚體與目的片段間有效分離,磁珠篩選通過不同比例PEG濃度將小片段完整去除,兩種方案都可以有效的將接頭二聚體去除,保證后期測序數據的高質量;第三,EA-M法通過末端修復以及加“A”反應,連接合并成一步,大大縮短了步驟,從而提高DNA回收效率,實現提高最終文庫得率的目的,WB-M法通過將DNA固定在磁珠上的方法減少DNA的被轉移次數,從而提高最終文庫總量,兩種方法分別從減少步驟和減少DNA轉移次數的角度提高了文庫得率,從而實現10~50 ng微量DNA起始樣品的RRBS文庫構建;最后,分別通過2種方法的穩定性實驗,可以看到2種方法的可重復性很高,成功率基本達到100%,并且針對同一個DNA樣本的重復性可以達到CV<0.05的要求,表明這2種方法構建RRBS文庫穩定性較好,并且WB-M因全程使用磁珠純化,適用于液體工作站上使用,在有效提高建庫規模化的同時,有效減少人工參與帶來的建庫誤差。
本研究中最關鍵的因素在于DNA起始量低,所以根據不同的DNA起始量,研發了2種方案,其中EA-M可以針對50~100 ng的微量DNA樣品的RRBS文庫構建,WB-M可以針對10~50 ng 的微量DNA樣品的RRBS文庫構建,然而WB-M方案針對50~100 ng DNA出發量時會有更好的效果。原因如下:(1)經過2個方案的比較發現,減少DNA轉移次數可以大大降低DNA在建庫中的損耗及損傷,DNA被固定在磁珠上對DNA自身也具有一定保護作用,所以WB-M方案可以有效保證酶切末端,減少在建庫過程中酶切末端的損傷。(2)Unique Reads Ratio(唯一不重復Reads比例)是二代測序數據質控中的一個重要指標,而影響該指標的因素主要是DNA起始量和文庫構建中PCR的循環數。本課題研究的微量DNA RRBS文庫構建方案,起始DNA樣本量少成為影響該指標的制約因素,因此如何有效減少文庫構建中PCR循環數,成為本研究中的關鍵因素。WB-M可以有效減少DNA損耗,使最終減少PCR循環數變成可能,在后續實驗中,發現50 ng DNA起始樣本量可以將最終PCR循環次數降低至10次,10 ng DNA起始可有效降低PCR循環數至14次,大大提高測序數據的有效性。經過2種方案的比較發現,WB-M方法在構建微量DNA RRBS文庫時,比CO-M和EA-M更加適合,但EA-M在文庫構建時間上有一定優勢,因為減少了純化和反應程序,所以EA-M法構建RRBS文庫比其他2種方法減少約1/3時間。另一方面,從經濟角度來看,EA-M的成本是CO-M的70%左右,WB-M的成本是CO-M的77%左右,所以在進行100 ng 左右DNA起始RRBS文庫時,從時間和經濟的角度上,推薦使用EA-M法進行文庫構建,但從質量上來看,WB-M法則更為優秀。
本研究中因偏重于人來源的微量樣本研究(例如:石蠟切片,顯微微切割等),所以只使用了人來源的gDNA樣本,后期驗證WB-M(因WB-M方案制備的文庫質量更優于EA-M方案,且在實際應用中WB-M方案更適合檢測技術發展方向,所以本驗證實驗只驗證了WB-M方案。)也只使用了人來源樣本,并通過信息學分析可看到WB-M在同時處理常量樣本和微量樣本時,Unique Reads均可達到65%以上,并且可發現的CpG context中C堿基甲基化比例也保持高度一致。本研究中提及的WB-M已成功應用于顯微微切割樣本的處理。目前還未應用于FFPE樣本的處理,從WB-M方案來看,可以適用于FFPE樣本的處理,但在進行MspI酶切之后,應使用UDG和FpG酶對FFPE來源DNA進行DNA修復,從而提高文庫構建的成功率。隨著二代測序技術的發展,單細胞測序也廣泛應用到醫療及診斷領域,如PGD,CTCs等,本課題研究中推薦的WB-M方案也可應用于單細胞領域,通過單細胞擴增技術將單個細胞DNA少量擴增后,使用WB-M方案進行微量DNA樣品的RRBS文庫構建,可以獲得高質量的RRBS文庫,為后續測序及生物信息分析打下堅實基礎。
References)
[1]GOLDBERG A D, ALLIS C D, BERNSTEIN E. Epigenetics:a landscape takes shape[J]. Cell, 2007, 128(4):635-638.DOI:10.1016/j.cell.2007.02.006.
[2]BIRD A P. CpG islands as gene markers in the vertebrate nucleus[J]. Trends in Genetics, 1987, 3(12):342-347.DOI∶10.1016/0168-9525(87)90294-0.
[3]BIRD A. DNA methylation patterns and epigenetic memory[J]. Genes & Development, 2002, 16(1):6-21.DOI:10.1101/gad.947102.
[4]黃慶,郭穎, 府偉靈.人類表觀基因組計劃[J].生命的化學, 2004, 24 (2):101-103.
HUANG Qing, GUO Yin, FU Weiling.Human epigenome project [J]. Chemistry of Life, 2004,24(2):101-103.
[5]ESTELLER M, ALMOUZNI G. How epigenetics integrates nuclear functions. Workshop on epigenetics and chromatin:transcriptional regμLation and beyond[J]. EMBO Reports, 2005, 6 (7):624-628.DOI:10.1038/sj.embor.7400456.
[6]GARDINER G M, FROMMER M. CpG islands in vertebrate genomes[J]. Journal of MolecμLar Biology, 1987, 196(2): 261-282. DOI∶10.1016/0022-2836(87)90689-9.
[7]TAKAI D, JONES P A. Comprehensive analysis of CpG islands inhuman chromosomes 21 and 22[J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99(6):3740-3745.DOI: 10.1073/pnas.052410099.
[8]WANG Y, LEUNG F C. An evaluation of new criteria for CpG islandsin the human genome as gene markers[J]. Bioinformatics, 2004, 20(7):1170-1177.DOI:10.1093/bioinformatics/bth059.
[9]LARSEN F, GUNDERSEN G, LOPEZ R, et al. CpG islands as gene markersin the human genome[J]. Genomics, 1992, 13(4):1095-1107.DOI:10.1016/0888-7543(92)90024-M.
[10]HANSEN R S, STOGER R, WIJMENGA C, et al. Escape from genesilencing in ICF syndrome:evidence for advanced replication timeas a major determinant[J]. Human MolecμLar Genetics, 2000, 9(18):2575-2587.DOI:10.1093/hmg/9.18.2575.
[11]YAMADA Y, WATANABE H, MIURA F, et al. A comprehensive analysisof allelic methylation status of CpG islands on human chromosome21q[J]. Genome Research, 2004, 14(2):247-266.DOI:10.1101/gr.1351604.
[12]HANAHAN D, WEINBERG R A. The hallmarks of cancer[J]. Cell, 2000,100(1):57-70.DOI:10.1016/S0092-8674(00)81683-9.
[13]SuLTAN M,SCHuLZ M H,RICHARD H,et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome[J].Science,2008,321(5891) :956-960.DOI:10.1126/science.1160342.
[14]SCHUSTER S C. Next-generation sequencing transforms today’s biology[J]. Nature Methods,2008,5(1) :16-18.DOI:10.1038/nmeth1156.
[15]COKUS S J, FENG S, ZHANG X, et al.Shotgun bisμLphate sequencing of theArabidopsisgenome reveals DNA methylation patterning[J]. Nature, 2008,452(7184):215-219.DOI:10.1038/nature06745.
[16]LI N,YE M,LI Y,et al. Whole genome DNA methylation analysis based on high throughput sequencing technology[J].Methods,2010,52(3) :203-212.DOI:10.1016/j.ymeth.2010.04.009.
[17]YAN H H, KIKUCHI S, NEUMANN P, et al. Genome-wide mapping of cytosine methylation revealed dynamic DNA methylation patterns associated with genes and centromeres in rice[J]. Plant Journal,2010,63(3) :353-365.DOI:10.1111/j.1365-313X.2010.04246.x.
[18]XIANG H, ZHU J, CHEN Q, et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map[J]. Nature Biotechnology, 2010, 28(5) :516-520.DOI:10.1038/nbt.1626.
[19]LISTER R, PELLZZOLA M, DOWEN RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences[J]. Nature, 2009, 462(7271):315-322.DOI:10.1038/nature08514.
[20]TURCOT V, BOUCHARD L, FAUCHER G, et al. DPP4 gene DNA methylation in the omentum is associated with its gene expression and plasma lipid profile in severe obesity[J]. Obesity,2011, 19(2):388-95.DOI:10.1038/oby.2010.198.
[21]LI Y, ZHU J, TIAN G, et al. The DNA methylome of human peripheral blood mononuclear cells[J]. PLoS Biology, 2010,8(11):e1000533.DOI:10.1371/journal.pbio.1000533.
[22]MEISSNER A, GNIRKE A, BELL G W, et al. Reduced representation Bisulfite sequencing for comparative high-resolution DNA methylation analysis[J]. Nucleic Acids Research, 2005,33(18):5868-5877.DOI:10.1093/nar/gki901.
[23]SMITH Z D, GU H C, BOCK C, et al. High-throughput Bisulfite sequencing in mammalian genomes[J]. Methods,2009, 48(3):226-232.DOI:10.1016/j.ymeth.2009.05.003.
[24]GU H C, Bock C, MIKKELSEN T S, et al. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution[J]. Nature Methods, 2010, 7(2):133-136. DOI:10.1038/nmeth.1414.
[25]WANG L, SUN J, WU H, et al. Systematic assessment of reduced representation Bisulfite sequencing to human blood samples:A promising method for large-sample-scale epigenomic studies[J]. Journal of Biotechnology,2012, 157(1):1-6.DOI:10.1016/j.jbiotec.2011.06.034.
[26]WANG J, XIA Y, LI L, et al. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation Bisulfite sequencing[J]. BMC Genomics,2013, 14(1):1-12.DOI:10.1186/1471-2164-14-11.
[27]CHRISTOPH B, ELENI M T, ARIE B B, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies[J]. Nature biotechnology,2010, 28(10):1106-1114.DOI:10.1038/nbt.1681.
[28]GU H,SMITH Z D,BOCK C, et al. Preparation of reduced representation Bisulfite sequencing librariesfor genome-scale DNA methylation profiling[J]. Nature Protocol, 2011, 6(4):468-481.DOI:10.1038/nprot.2010.190.
[29]MAXIMILIAAN S, ANJA S, LOBS A K, et al. Laser capture microdissection-reduced representation Bisulfite sequencing (LCM-RRBS)maps changes in DNA methylation associated with gonadectomy-induced adrenocortical neoplasia in the mouse[J]. Nucleic Acids Research, 2013, 41(11 ):249-274.DOI:10.1093/nar/gkt230.
An μLtra-low-input RRBS (Reduced Representation Bisulfite Sequencing) library preparation method
WEI Dongkai1,2, NAN Peng2, XU Kangping1, QIU Feng1, 3*
(1.BasePairBio-TechnologyCo.Ltd,JiangSuSuzhou215123,China;2.SchoolofLifeSciencesFudanUniversity,Shanghai200438,China;3.ShanghaicenterforBioinformationtechnology,Shanghai201203,China)
RRBS (Reduced Representation Bisulfite Sequencing) is an effective method for DNA methylation study. Library preparation is one of the most critical steps of RRBS experiments. In conventional RRBS library preparation, it usually needs 1 ug or more starting DNA because of less enzyme digested products and losses in various experiment steps. Meanwhile, many clinical samples from patients, such as frozen puncture tissue, FFPE, microdissection, etc., there is only a limited amount of extracted DNA less than 100 ng, and they don’t fit the requirement of the conventional RRBS library preparation, which greatly limits the application field of RRBS technology. This research studies and designs RRBS library preparation using μLtra-low-input DNA samples (<100 ng), mainly by optimizing MspI digestion condition, DNA fragment size selection, reduction of reaction steps and DNA transfer steps in library preparation, which greatly improve the DNA recovery. We set up two effective μLtra-low-input RRBS library preparation methods (EA-Method and WB-Method). The EA-Method is more cost effective, fast, and efficient than WB-Method when DNA input amount around 100 ng, but the library quality is slightly lower than the WB-Method; The WB-Method has higher quality performance when using 10 ng DNA. In order to test the reliability, we construct 3 groups of RRBS libraries (1ug DNA input and 10ng DNA input) from three volunteer samples (whole blood and mouth swab) by WB-Method. The prepared libraries are sequenced on Illumina Hiseq platform. The compatible resμLts are obtained comparing the μLtra-low-input and conventional RRBS method by analysis of detected C bases in the CpG, CHG, CHH methylation rate. Nevertheless, the μLtra-low-input RRBS method can greatly reduce DNA loss and damage during library preparation, and it will be a wider application of RRBS technology to different type samples and promote broader research field of RRBS technology effectively.
NGS; RRBS; Reduced Representation Bisulfite Sequencing; EA-M; WB-M
2016-09-03;
2016-09-20.
魏冬凱,男,碩士研究生,研究方向:高通量測序技術及應用;E-mail:dkwei@basepair.cn.
*通信作者:裘鋒,男,研究員,研究方向:高通量測序及液態活檢; E-mail:qiufeng@scbit.org.
10.3969/j.issn.1672-5565.2017.01.201609001
Q78
A
1672-5565(2017)01-033-13
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
