Caroli病伴腎消耗病1例報(bào)告并文獻(xiàn)復(fù)習(xí)
2017-05-13 08:42:46歐陽雨晴陸海英徐小元
臨床肝膽病雜志 2017年5期
歐陽雨晴, 陸海英, 徐小元
(北京大學(xué)第一醫(yī)院 感染疾病科, 北京 100034)
Caroli病伴腎消耗病1例報(bào)告并文獻(xiàn)復(fù)習(xí)
歐陽雨晴, 陸海英, 徐小元
(北京大學(xué)第一醫(yī)院 感染疾病科, 北京 100034)
Caroli病; 腎消耗病; 病例報(bào)告
Caroli病是一種先天性多發(fā)節(jié)段性的肝內(nèi)膽道擴(kuò)張合并先天性肝纖維化的疾病,臨床表現(xiàn)多樣,主要通過影像學(xué)表現(xiàn)診斷。Caroli病常并發(fā)多囊腎,但并發(fā)青少年腎消耗病(nephronophthisis,NPHP)的病例較為少見。現(xiàn)將1例Caroli病并發(fā)NPHP的患者報(bào)道如下。
1 病例資料
患者女性,18歲,因“反復(fù)水腫11個(gè)月,肝功能異常7個(gè)月”于2016年10月24日入本院。患者11個(gè)月前無明顯誘因出現(xiàn)雙下肢水腫,未予診治。7個(gè)月前患者于發(fā)熱、干咳后水腫加重,由顏面部逐漸蔓延至全身,伴腰痛,當(dāng)?shù)蒯t(yī)院查尿蛋白陽性(+),尿潛血陽性(+++),尿蛋白定量0.62 g/24 h;肝功能:AST 68 U/L,Alb 16.7 g/L,TBil 66.3 μmol/L,DBil 23.44 μmol/L,肌酐99 μmol/L,IgA 6.03 g/L,IgM 3.54 g/L,補(bǔ)體C3 0.396 g/L,補(bǔ)體C4 0.086 g/L;腹部超聲示:肝臟彌漫性病變,脾大,雙腎彌漫性病變,考慮“急性腎小球腎炎”。予頭孢呋辛、人血白蛋白治療后,水腫、發(fā)熱癥狀緩解。后患者反復(fù)出現(xiàn)水腫3次,均經(jīng)對癥治療癥狀緩解。2周前患者出現(xiàn)發(fā)熱、干咳,AST 93.6 U/L,ALT 44.7 U/L,Alb 17.9 g/L,TBil 44.2 μmol/L,DBil 13.4 μmol/L,擬膽堿酯酶 4420.1 U/L,肌酐124.9 μmol/L;尿蛋白陽性(++)、尿潛血陽性(+++),尿微量白蛋白245.0 mg/L。予阿奇霉素、感冒靈治療后退熱,但出現(xiàn)全身水腫伴腹脹、尿量減少,為進(jìn)一步診治轉(zhuǎn)入本院。既往史:患者7歲時(shí)出現(xiàn)肉眼血尿、肝脾增大;原發(fā)性閉經(jīng),診斷為“多囊卵巢綜合征(polycystic ovarian syndrome,PCOS)”2年。入院查體:血壓108 mm Hg/58 mm Hg,全身皮膚黏膜水腫,皮膚、鞏膜無黃染,無肝掌及蜘蛛痣。腹部膨隆,移動(dòng)性濁音陽性,肝脾肋下未觸及,雙下肢中度可凹性水腫。肝功能:ALT 33 U/L,AST 68 U/L,Alb 11.7 g/L,ALP 71 U/L,GGT 18 U/L,TBil 35.7 μmol/L,DBil 8.6 μmol/L;腎功能:血肌酐 96.8 μmol/L,尿素氮 10.67 mmol/L,內(nèi)生肌酐清除率 63.24 ml/min,血鉀3.63 mmol/L,血鈉142 mmol/L;凝血功能:PT 15.6 s, 血清前白蛋白 57%,國際標(biāo)準(zhǔn)化比值1.54;血常規(guī):WBC 3.8×109/L,Hb 87 g/L,PLT 88×109/L;尿常規(guī):尿蛋白陽性(++)、潛血陽性(+++);尿蛋白電泳:小分子蛋白0,白蛋白86.7%,大分子蛋白13.3%;尿紅細(xì)胞位相:腎性血尿;24 h尿蛋白定量4.52 g/L,尿鈉551.2 mmol/24 h,尿鉀63.6 mmol/24 h;尿滲透壓375 mOsm/kg,血滲透壓290 mOsm/kg。病毒性肝炎抗體陰性,自身免疫抗體陰性。性激素:睪酮0.83 ng/ml,其他激素水平正常。腹部超聲:肝硬化,肝內(nèi)多發(fā)囊性病變,脾臟增厚,雙腎增大、彌漫性病變、皮髓質(zhì)顯示不清,大量腹水。肝臟彈性值為69.1 kPa。腹部增強(qiáng)CT(圖1):肝內(nèi)膽管囊性擴(kuò)張,肝硬化,門靜脈高壓,門體分流,脾大,腹水,皮下組織水腫,胸腔積液,考慮Caroli病。腎臟MRI、胸部和頭顱CT平掃及眼底檢查未見異常。基因檢測:NPHP2、CC2D2A基因突變。臨床診斷:Caroli病失代償期肝硬化,門靜脈高壓,腹水;NPHP,慢性腎炎綜合征,慢性腎臟病2期;PCOS,原發(fā)性閉經(jīng)。入院后予白蛋白、血漿輸注對癥支持、利尿消腫、保肝治療以及洛汀新降尿蛋白治療,患者病情緩解出院。
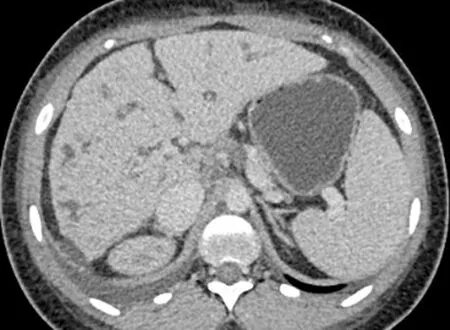
圖1 腹部增強(qiáng)CT 肝內(nèi)膽管囊性擴(kuò)張,中心可見對比增強(qiáng)的門靜脈分支
2 討論
Caroli病是指膽道擴(kuò)張合并先天性肝纖維化的疾病,多表現(xiàn)為常染色體隱性遺傳,常同時(shí)合并常染色體隱性遺傳性多囊腎[1-4]。Caroli病的發(fā)病機(jī)制尚未完全明確,有研究[5-7]表明導(dǎo)致常染色體隱性遺傳多囊腎病的基因PKHD1編碼蛋白在肝、腎均有表達(dá),定位在纖毛,纖毛對肝及膽道系統(tǒng)中細(xì)胞增殖和板狀細(xì)胞極性的維持起著重要作用,在較大肝內(nèi)膽管的形成中,導(dǎo)管板重塑的停滯和紊亂會(huì)導(dǎo)致肝內(nèi)膽管的擴(kuò)張和肝纖維化。
Caroli病主要表現(xiàn)為膽管擴(kuò)張導(dǎo)致的膽汁淤積、肝內(nèi)膽石癥以及細(xì)菌性膽管炎、菌血癥、肝膿腫。一部分患者可表現(xiàn)為門靜脈高壓及相關(guān)并發(fā)癥,例如腹水、食管胃底靜脈曲張破裂出血,少數(shù)患者僅表現(xiàn)為間斷腹痛。皮膚瘙癢和肝脾大是常見體征,實(shí)驗(yàn)室檢查主要表現(xiàn)為ALP、DBil水平升高,肝臟合成功能隨疾病進(jìn)展逐漸惡化。Caroli病的診斷依賴于影像學(xué)檢查,表現(xiàn)為膽道擴(kuò)張和肝內(nèi)大膽管的不規(guī)則囊樣擴(kuò)張,但膽總管正常。采用超聲、經(jīng)內(nèi)鏡逆行胰膽管造影、磁共振胰膽管造影等檢查方法都可明確診斷,所謂“中心點(diǎn)征”是指擴(kuò)張的肝內(nèi)膽管完全包繞對比增強(qiáng)的門靜脈小分支[8]。Caroli病的治療方法主要為支持對癥治療和個(gè)體化治療。膽管炎和敗血癥患者需抗感染、取石治療;慢性膽汁淤積患者需要補(bǔ)充脂溶性維生素;食管胃底靜脈曲張患者需預(yù)防性應(yīng)用非選擇性β受體阻滯劑;選擇性分流術(shù)能緩解門靜脈高壓。治療過程中發(fā)生不能解釋的病癥惡化或新發(fā)膽道狹窄都應(yīng)考慮膽管癌的可能。對于反復(fù)膽道感染,尤其是出現(xiàn)門靜脈高壓相關(guān)并發(fā)癥的患者,應(yīng)進(jìn)行肝移植,合并終末期腎病的患者可行肝腎聯(lián)合移植[9-10]。
Caroli病并發(fā)NPHP的病例較為少見,是常染色體隱性病[11],NPHP致病基因表達(dá)的蛋白主要分布于初級纖毛、基粒以及中心體上,NPHP基因突變可引起纖毛功能異常,腎小管細(xì)胞無法感知液體流動(dòng)導(dǎo)致組織生長失調(diào)、腎囊腫形成。其最初的臨床表現(xiàn)是由于腎小管濃縮功能和保鈉功能障礙而造成多尿、煩渴、尿滲透壓低。鈉離子丟失會(huì)造成血容量不足、低鈉血癥。NPHP常會(huì)在30歲前進(jìn)展到終末期腎病,腎衰竭進(jìn)展速度取決于疾病類型、基因缺陷程度。10%~20%的NPHP患者可伴有腎外表現(xiàn),例如錐形骺、肝脾大、門靜脈纖維化、內(nèi)臟轉(zhuǎn)位和室間隔缺損等。NPHP的臨床特點(diǎn)包括多尿,進(jìn)展性慢性腎病,超聲示腎臟回聲增強(qiáng)、皮髓分界不清;致病基因檢測陽性或腎臟病理表現(xiàn)為腎小管基底膜增厚的慢性腎小管間質(zhì)改變可以證實(shí)NPHP的診斷[12-14]。NPHP的治療主要為糾正水、電解質(zhì)紊亂的支持治療,腎移植是終末期腎病患者的首選治療方法。
本例患者為青年女性,存在多系統(tǒng)及器官病變。肝病方面,患者主要表現(xiàn)為轉(zhuǎn)氨酶和膽紅素輕度升高,低白蛋白血癥及腹水。入院后查各型病毒性肝炎標(biāo)志物未見異常,自身免疫抗體陰性,可排除病毒性肝炎和自身免疫性肝病。患者無明確用藥及飲酒史,不考慮藥物性肝損傷及酒精性肝病可能。腹部增強(qiáng)CT提示肝內(nèi)多發(fā)分支囊狀低密度灶,內(nèi)部可見靜脈及門靜脈分支,呈“中心點(diǎn)征”改變,伴肝硬化,符合Caroli病影像學(xué)表現(xiàn),故Caroli病診斷明確。Caroli病常合并多囊腎病,但本例患者入院后腎臟MR除外合并腎囊腫可能。該患者臨床表現(xiàn)為多尿、血容量不足、血壓偏低等,慢性腎功能不全,尿滲透壓降低、尿鈉增加等腎小管濃縮功能障礙表現(xiàn),腎臟超聲示腎臟回聲增強(qiáng)、皮髓質(zhì)分界不清,符合NPHP的臨床表現(xiàn);基因檢測結(jié)果提示NPHP2和CC2D2A基因的多個(gè)位點(diǎn)突變,其中NPHP2基因突變支持NPHP的診斷,故Caroli病合并NPHP診斷成立。此外,患者有大量蛋白尿、低白蛋白血癥、反復(fù)水腫、血尿等表現(xiàn),尿蛋白電泳以大分子蛋白為主,紅細(xì)胞位相提示腎性血尿,考慮同時(shí)存在腎小球疾病可能,結(jié)合患者血IgA升高,補(bǔ)體C3、C4減低,繼發(fā)性腎小球腎炎相關(guān)檢查陰性,不除外肝功能異常導(dǎo)致IgA清除能力減弱引發(fā)繼發(fā)性IgA腎病可能,鑒于患者為失代償期肝硬化,凝血功能差,暫無條件完善病理檢查明確診斷。婦科方面患者表現(xiàn)為PCOS、原發(fā)性閉經(jīng)。有報(bào)道[15-16]多囊腎病可合并PCOS,但該患者無多囊腎病。卵巢分泌過多的雄激素是導(dǎo)致PCOS的常見原因,患者入院后查雄激素輕度升高,故不除外高雄激素血癥導(dǎo)致PCOS的可能, 患者原發(fā)性閉經(jīng)可能與PCOS相關(guān)。入院后予補(bǔ)充人血白蛋白、輸注新鮮冰凍血漿擴(kuò)容、小劑量利尿等支持治療,患者體質(zhì)量下降15 kg,腹水及全身水腫癥狀消失,Alb逐漸升高至29.4 mg/L,但患者出現(xiàn)尿蛋白增多,加用血管緊張素轉(zhuǎn)化酶抑制劑類藥物降尿蛋白治療,尿蛋白定量由4.52 g/24 h降至1.6 g/24 h。因患者肝內(nèi)膽管囊腫分布于雙葉伴肝硬化,合并門靜脈高壓、門體分流建立,肝功能Child-Pugh評分B級,建議患者盡早行肝移植手術(shù)改善預(yù)后。
通過本病例的診治及相關(guān)文獻(xiàn)的復(fù)習(xí),獲得以下經(jīng)驗(yàn)和體會(huì):對于年輕,肝損傷較輕,同時(shí)存在肝硬化、門靜脈高壓表現(xiàn)的患者,臨床診斷應(yīng)考慮Caroli病等一些少見病的可能。影像學(xué)檢查對Caroli病的診斷十分重要,但須醫(yī)師具備豐富的影像學(xué)知識和經(jīng)驗(yàn)。此外,該病的治療比較棘手,低蛋白血癥與蛋白尿互為因果,治療上相互矛盾,預(yù)后差,缺乏特效方法,肝移植或肝腎聯(lián)合移植是改善預(yù)后的最佳途徑。
[1] LIANG JJ, KAMATH PS. Caroli syndrome[J]. Mayo Clin Proc, 2013, 88(6): e59. [2] PARK E, LEE JM, AHN YH, et al. Hepatorenal fibrocystic diseases in children[J]. Pediatr Nephrol, 2016, 31(1): 113-119.
[3] SWEENEY WE, AVNER ED. Polycystic kidney disease, autosomal recessive[EB/OL]. Gene Rev, 2001. https://www.ncbi.nlm.nih.gov/pubmed/20301501
[4] OBUSEZ EC, UDAYASANKAR U. Autosomal recessive polycystic kidney disease with Caroli syndrome[J]. J Urol, 2015, 193(2): 679-680.
[5] WARD CJ, HOGAN MC, ROSSETTI S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein[J]. 2002, 30(3): 259-269.
[6] COURCET JB, MINELLO A, PRIEUR F, et al. Compound heterozygous PKHD1 variants cause a wide spectrum of ductal plate malformations[J]. Am J Med Genet A, 2015, 167(12): 3046-3053.
[7] HAO X, LIU S, DONG Q, et al. Whole exome sequencing identifies recessive PKHD1 mutations in a Chinese twin family with Caroli disease[J]. PLoS One, 2014, 9(4): e92661.
[8] PERRICONE G, VANZULLI A. Education and imaging. Hepatology: "central dot sign" of Caroli syndrome[J]. J Gastroenterol Hepatol, 2015, 30(2): 234.
[9] TELEGA G, CRONIN D, AVNER ED. New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications[J]. Pediatr Transplant, 2013, 17(4): 328-335.
[10] WEN JW, FURTH SL, RUEBNER RL. Kidney and liver transplantation in children with fibrocystic liver-kidney disease: data from the US Scientific Registry of Transplant Recipients: 1990-2010[J]. Pediatr Transplant, 2014, 18(7): 726-732.
[11] STOKMAN M, LILIEN M, KNOERS N. Nephronophthisis[EB/OL]. Gene Reviews, 2016. https://www.ncbi.nlm.nih.gov/pubmed/27336129
[12] HAGHIGHI A, SAVAJ S, HAGHIGHI-KAKHKI H, et al. Identification of an NPHP1 deletion causing adult form of nephronophthisis[J]. Ir J Med Sci, 2016, 185(3): 589-595.
[13] KANG HG, LEE HK, AHN YH, et al. Targeted exome sequencing resolves allelic and the genetic heterogeneity in the genetic diagnosis of nephronophthisis-related ciliopathy[J]. Exp Mol Med, 2016, 48(8): e251.
[14] LEE JM, AHN YH, KANG HG, et al. Nephronophthisis 13: implications of its association with Caroli disease and altered intracellular localization of WDR19 in the kidney[J]. Pediatr Nephrol, 2015, 30(9): 1451-1458.
[15] PEREGRIN AI, URBANEJA LM, QUIJADA TD. Polycystic ovarian syndrome in a woman with polycystic kidney disease[J]. Eur J Obstet Gynecol Reprod Biol, 2008, 140(2): 282-283.
[16] SEGASOTHY M, NORAZLINA MY, ONG PH, et al. Polycystic kidney disease associated with polycystic ovarian syndrome[J]. Nephron, 1992, 62(4): 482-483.
引證本文:OUYANG YQ, LU HY, XU XY. Caroli disease with nephronophthisis: a case report and literature review[J]. J Clin Hepatol, 2017, 33(5): 946-948. (in Chinese) 歐陽雨晴, 陸海英, 徐小元. Caroli病伴腎消耗病1例報(bào)告并文獻(xiàn)復(fù)習(xí)[J]. 臨床肝膽病雜志, 2017, 33(5): 946-948.
(本文編輯:邢翔宇)
Caroli disease with nephronophthisis: a case report and literature review
OUYANGYuqing,LUHaiying,XUXiaoyuan.
(DepartmentofInfectiousDiseases,PekingUniversityFirstHospital,Beijing100034,China)
Caroli disease; nephronophthisis; case reports
10.3969/j.issn.1001-5256.2017.05.033
2016-11-29;
2016-12-18。
國家十二五科技重大專項(xiàng)資助項(xiàng)目(2012ZX10002003)
歐陽雨晴(1991-),女,在讀博士,主要從事肝病方面的研究。
陸海英,電子信箱:luhaiying00@126.com。
R575.7; R692
B
1001-5256(2017)05-0946-03
