麻黃堿、偽麻黃堿及(1S,2S)-β-氯代甲基苯丙胺、(1R,2S)-β-氯代甲基苯丙胺的分析方法研究
2017-10-11 02:36:45錢振華李靜花鎮東
中國司法鑒定 2017年5期
關鍵詞:分析
錢振華,李靜,花鎮東
(1.公安部物證鑒定中心,北京100038;2.公安部禁毒情報技術中心,北京100193)
鑒定科學ResearchPaper
麻黃堿、偽麻黃堿及(1S,2S)-β-氯代甲基苯丙胺、(1R,2S)-β-氯代甲基苯丙胺的分析方法研究
錢振華1,李靜2,花鎮東2
(1.公安部物證鑒定中心,北京100038;2.公安部禁毒情報技術中心,北京100193)
目的建立甲基苯丙胺制毒原料樣品中麻黃堿、偽麻黃堿及(1S,2S)-β-氯代甲基苯丙胺、(1R,2S)-β-氯代甲基苯丙胺的定性和定量分析方法。方法分別使用氣相色譜質譜聯用法(GC-MS)、液相色譜質譜聯用法(LC-MS)和液相色譜紫外檢測法(LC-UV)分析該類樣品,考察各方法的特點及適用范圍。結果GC-MS可以對麻黃堿和偽麻黃堿進行定性分析,最低檢測限為4.0 μg/mL,但進樣口的高溫會導致(1S,2S)-β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺轉化生成1,2-二甲基-3-苯基-氮丙啶。LC-MS及LC-UV分析則不存在這一問題,前者對麻黃堿、偽麻黃堿的最低檢測限為0.3 ng/mL,對(1S,2S)-β-氯代甲基苯丙胺、(1R,2S)-β-氯代甲基苯丙胺的最低檢測限為1.0 ng/mL;后者對麻黃堿和偽麻黃堿的最低檢測限為3.0μg/mL,對(1S,2S)-β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺的最低檢測限為5.0μg/mL。結論GC-MS僅適用于分析樣品中麻黃堿和偽麻黃堿,不能檢測樣品中的(1S,2S)-β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺。LC-MS可同時對樣品中的痕量麻黃堿、偽麻黃堿及(1S,2S)-β-氯代甲基苯丙胺、(1R,2S)-β-氯代甲基苯丙胺進行定性和定量分析,LC-UV則適用于上述物質的常量分析。
法醫毒物學;麻黃堿;偽麻黃堿;(1S,2S)-β-氯代甲基苯丙胺;(1R,2S)-β-氯代甲基苯丙胺
Abstract:ObjectiveTo establish methods for qualitative and quantitative analysis of ephedrine,pseudophedrine,(1S,2S)-β-Chloro-methamphetamine and(1R,2S)-β-Chloro-methamphetamine in precursor of methamphetamine seizures.MethodsGC/MS,LC-MS and LC-UV methods were developed for the analysis of these substances.Their features and scopes were compared and discussed.ResultsEphedrine and pseudophedrine presented good resolution using GC/MS,and LODs were 4.0 μg/mL.However,the transformation of(1S,2S)-β-Chloro-methamphetamine and(1R,2S)-β-Chloromethamphetamine to 1,2-dimethyl-3-phenylaziridine was observed in GC/MS analysis due to the high temperature of inlet. In contrast,such interference was entirely avoided using LC-MS or LC-UV.LODs of ephedrine and pseudophedrine were 0.3 ng/mL with LC-MS and 3.0 μg/mL with LC-UV.LODs of(1S,2S)-β-Chloro-methamphetamine and(1R,2S)-β-Chloro-methamphetamine were 1.0 ng/mL with LC-MS and 5.0 μg/mL with LC-UV.ConclusionGC/MS was fit for the identification of ephedrine and pseudophedrine,but not for(1S,2S)-β-Chloro-methamphetamine and(1R,2S)-β-Chloro-methamphetamine.LC-MS showed excellent performance in the detection of trace ephedrine,pseudophedrine,(1S,2S)-β-Chloro-methamphetamine and(1R,2S)-β-Chloro-methamphetamine,while LC-UV provided an alternative for routine quantitative analysis.
Keywords:forensic toxicology;ephedrine;pseudophedrine;(1S,2S)-β-Chloro-methamphetamine;(1R,2S)-β-Chloromethamphetamine
近年來,在國內外毒品形勢影響下,我國吸毒人數呈幾何式增長,其中甲基苯丙胺(冰毒)濫用人數最多,非法制販問題日趨嚴峻。根據相關案件的情況,目前我國地下加工廠主要以麻黃堿/偽麻黃堿為原料,通過催化加氫法合成甲基苯丙胺。催化加氫法的第一步是將麻黃堿/偽麻黃堿制成β-氯代甲基苯丙胺(俗稱氯代麻黃堿、氯麻黃堿、熟麻),第二步是將β-氯代甲基苯丙胺還原成冰毒[1-3](圖1)。由于β-氯代甲基苯丙胺還未列入管制,一些麻黃堿加工廠開始生產β-氯代甲基苯丙胺,以逃避公安機關的打擊,同時也可以減少下游甲基苯丙胺加工廠的生產時間,降低其被發現的風險。近來全國各地特別是福建禁毒部門在案件中繳獲大量麻黃堿和β-氯代甲基苯丙胺。自天然麻黃草和含麻黃堿復方制劑的管制加強以后,不法分子開始使用溴代苯丙酮為原料,通過化學合成法制造麻黃堿。這類合成方法得到的產物是一個混合物,包括麻黃堿(約90%)和偽麻黃堿(約10%)。使用該混合物繼續生產β-氯代甲基苯丙胺,也同樣會得到(1S,2S)-β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺兩種產物,且兩者的理論比例約為9∶1。因此,準確測定此類案件的現場繳獲樣品的成分及含量,對判斷加工廠的性質及其在制毒環節中的作用具有重要的意義。本文考察了氣相色譜質譜聯用法(GCMS)、液相色譜質譜聯用法(LC-MS)和液相色譜紫外檢測法(LC-UV)用于該類樣品分析時的特點,建立了基于LC-MS和LC-UV的定性和定量分析方法,并應用于對實際繳獲樣品的分析。

圖1 催化加氫法合成冰毒流程
1 材料與方法
1.1 儀器與試劑
QP-2010 Ultra氣相色譜質譜聯用儀(日本島津公司),TripleTOF 5600超快速液相色譜-四極桿飛行時間串聯質譜聯用儀(美國AB SCIEX公司),Aglient 1200高效液相色譜儀(美國安捷倫公司)。
麻黃堿鹽酸鹽、偽麻黃堿鹽酸鹽對照品(從繳獲樣品中提純,經核磁共振波譜儀確認純度大于99.5%)、(1R,2S)-β-氯代甲基苯丙胺鹽酸鹽、(1S,2S)-β-氯代甲基苯丙胺鹽酸鹽對照品(國家毒品實驗室合成,經核磁共振波譜儀確認純度大于98%),甲醇(色譜純,Merck公司),乙腈(色譜純,Merck公司),甲酸(色譜純,Sigma-Aldrich公司),磷酸(分析純,國藥集團),NaH2PO4(分析純,國藥集團),純水由Millipore純水儀制備。
1.2 標準溶液配制
精密稱取麻黃堿、偽麻黃堿、(1R,2S)-β-氯代甲基苯丙胺、(1S,2S)-β-氯代甲基苯丙胺鹽酸鹽對照品適量[含麻黃堿、偽麻黃堿、(1R,2S)-β-氯代甲基苯丙胺、(1S,2S)-β-氯代甲基苯丙胺堿型25mg],分別置于10 mL容量瓶中,加入甲醇溶解并稀釋至刻度,充分混勻,配制成濃度為2.5 mg/mL的貯備液。實驗時按所需濃度稀釋使用。
1.3 磷酸緩沖液配制
燒杯中加入約200 mL超純水,再量取85%濃磷酸2.0 mL并稱取NaH2PO42.2 g加入其中,用超純水稀釋至1 000 mL,攪拌至完全溶解,混合均勻后使用0.45 μm濾膜過濾,超聲后靜置待用。
1.4 GC-MS分析條件
色譜柱:Aglient DB-35MS石英毛細管柱(30 m ×0.25 mm×0.25 μm)。柱溫125℃,保持10 min,以5℃/min升至140℃,再以40℃/min升至300℃,保持3min;載氣為He,流速1 mL/min;分流進樣,進樣量1 μL,分流比20∶1;進樣口溫度280℃。EI電離模式,能量70eV,離子源溫度230℃,接口溫度250℃,掃描范圍m/z 35~500。
1.5 LC-MS分析條件
液相部分:色譜柱:Waters ACQUITY UPLC CSH C18(1.7μm,100mm×2.1mm)。A相為0.1%甲酸水溶液,B相為乙腈,梯度洗脫(表1)。流速0.4mL/min,樣品池溫度15℃,進樣量1 μL。

表1 液相梯度洗脫條件
質譜部分:DuoSpray離子源,ESI+模式,離子源溫度600℃,噴霧電壓5500 V,霧化氣50 Psi,輔助加熱氣50 Psi,氣簾氣30 Psi,去簇電壓80 V。TOF全掃描模式,掃描范圍m/z 100~1000;子離子掃描模式,母離子質量166.1226和184.0888,碰撞能量35±15 V,掃描范圍m/z 50~800。
1.6 LC-UV分析條件
色譜柱:AglientEclipsePlusC18色譜柱(3.5μm,100mm×4.6mm)。A相為磷酸緩沖液,B相為乙腈,梯度洗脫(表2)。流速1.5mL/min,柱溫箱35℃,進樣量5μL,二極管陣列檢測器,檢測波長210nm。

表2 液相梯度洗脫程序
1.7 樣品處理
樣品溶于甲醇,配制成所需濃度溶液,0.22 μm濾膜過濾后供分析。
2 結果與討論
2.1 GC-MS分析
國內外對GC-MS分析麻黃堿和偽麻黃堿早有研究,由于麻黃堿和偽麻黃堿為堿性化合物,易和色譜柱硅膠表面存在的酸性硅醇基發生相互作用,導致峰形拖尾,故可對樣品進行萃取或者將樣品溶解在含10 mmol/L氫氧化鈉的甲醇溶液中,改善峰形。雖然麻黃堿和偽麻黃堿是一對光學異構體,但使用本實驗條件,麻黃堿和偽麻黃堿可實現基線分離,總離子流色譜圖和質譜圖見圖2。麻黃堿的保留時間為12.69 min,偽麻黃堿的保留時間為13.07 min,最低檢測限為4.0 μg/mL。國外一直將β-氯代甲基苯丙胺作為生產甲基苯丙胺過程中產生的特征雜質進行研究[4-11],發現由于β-氯代甲基苯丙胺穩定性較低,在高溫高壓下會發生分子內關環反應而生成1,2-二甲基-3-苯基-氮丙啶(1,2-dimethyl-3-phenylaziridine),使用氣相色譜儀或氣相色譜質譜聯用儀對β-氯代甲基苯丙胺進行分析時得到的是其轉化產物的譜圖,從而致使檢測結論出錯。本實驗證實了這一問題,還發現樣品濃度和進樣口襯管惰性程度都會影響β-氯代甲基苯丙胺的轉化。低濃度樣品的轉化率要高于高濃度樣品;襯管使用時間越長,吸附活化位點越多,轉化率也越高。因此,GC-MS僅適用于分析樣品中麻黃堿和偽麻黃堿,不能檢測樣品中的(1S,2S)-β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺。同理,GC也不適用于(1S,2S)-β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺的定量分析。
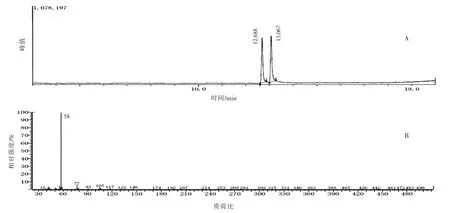
圖2 麻黃堿和偽麻黃堿GC-MS總離子流色譜圖(A)和質譜圖(B)
2.2 LC-MS分析
與GC-MS加熱發生在進樣階段不同,LC-MS的加熱發生在色譜分離后離子源內的離子化階段,因此不會造成β-氯代甲基苯丙胺的反應,影響樣品分析。利用LC-MS分析樣品中麻黃堿和偽麻黃堿的研究較多,但對(1S,2S)-β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺進行定性分析的報道較少[12],尚無報道對其進行定量分析。按1.5項條件進行全掃描分析,獲得提取離子色譜圖如圖3(提取離子[M+H]+,m/z=166.1221-166.1231)和圖4(提取離子[M+H]+,m/z=188.0883-188.0893)所示,麻黃堿的保留時間為1.54min,偽麻黃堿的保留時間為1.71min,(1S,2S)-β-氯代甲基苯丙胺的保留時間為2.82min,(1R,2S)-β-氯代甲基苯丙胺的保留時間為2.95min,達到基線分離,可同時對上述4種物質進行分析。
對麻黃堿/偽麻黃堿,一級質譜圖和以[M+H]+為母離子,碰撞誘導解離(CID)獲得的二級質譜圖見圖5。對(1S,2S)-β-氯代甲基苯丙胺/(1R,2S)-β-氯代甲基苯丙胺,一級質譜圖和以[M+H]+為母離子,碰撞誘導解離(CID)獲得的二級質譜圖見圖6。由于麻黃堿/偽麻黃堿和(1S,2S)-β-氯代甲基苯丙胺/(1R,2S)-β-氯代甲基苯丙胺的分子組成不同,因此一級質譜圖存在明顯區別,但兩者結構相似,碎裂方式也相似,因此二級質譜圖差異小,推測可能的碎裂途徑見圖7。
以0.1%的甲酸水溶液為稀釋液,分別配制系列濃度為0.3、1.0、3.0、10、30、100、300、1 000 ng/mL的麻黃堿鹽酸鹽、偽麻黃堿鹽酸鹽和(1S,2S)-β-氯代甲基苯丙胺鹽酸鹽、(1R,2S)-β-氯代甲基苯丙胺鹽酸鹽標準溶液進行分析。定性分析采取子離子掃描模式,麻黃堿和偽麻黃堿選擇m/z=166.1226、148.1121、133.0886、115.0542為定性離子,(1S,2S) -β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺選擇m/z=184.0888、148.1121、133.0886、115.0542為定性離子,以所有離子信噪比(S/N)大于3確定檢測限,麻黃堿和偽麻黃堿的檢出限均為0.3 ng/mL,而(1S,2S)-β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺的檢出限均為1.0 ng/mL。定量分析采取全掃描模式,以濃度x(ng/mL)為橫坐標,提取離子色譜峰面積y為縱坐標作線性回歸,得到麻黃堿的線性方程為y=177x+803,r2=0.9991,線性范圍3.0~300ng/mL;偽麻黃堿的線性方程為y=183x+796,r2=0.9993,線性范圍3.0~300ng/mL;(1S,2S)-β-氯代甲基苯丙胺的線性方程為y=494x+749,r2=0.9999,線性范圍3.0~300 ng/mL;(1R,2S)-β-氯代甲基苯丙胺的線性方程為y=512x+698,r2=0.9998,線性范圍3.0~300 ng/mL。
以上結果說明,LC-MS可對麻黃堿、偽麻黃堿、(1S,2S)-β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺進行準確的定性和定量。
2.3 LC-UV分析
LC-UV適用于樣品中常量組分的分析,按1.6項條件分析混合溶液,獲得的色譜圖如圖8所示。麻黃堿的保留時間為5.07 min,偽麻黃堿的保留時間為5.28 min,(1S,2S)-β-氯代甲基苯丙胺的保留時間為6.36 min,(1R,2S)-β-氯代甲基苯丙胺的保留時間為6.55 min,達到基線分離。
取麻黃堿、偽麻黃堿、(1R,2S)-β-氯代甲基苯丙胺、(1S,2S)-β-氯代甲基苯丙胺鹽酸鹽的標準貯備液適量,使用磷酸緩沖液進行稀釋,最終得到濃度為0.5、0.25、0.1、0.05、0.025、0.01 mg/mL系列標準溶液進行分析。以濃度x(mg/mL)為橫坐標,色譜峰面積y為縱坐標作線性回歸,得到四種物質的線性方程、線性范圍和檢出限(表3)。

圖3 麻黃堿和偽麻黃堿LC-MS提取離子色譜圖

圖4 (1S,2S)-β-氯代甲基苯丙胺和(1R,2S)-β-氯代甲基苯丙胺LC-MS提取離子色譜圖

圖5 麻黃堿/偽麻黃堿一級質譜圖(A)和二級質譜圖(B)
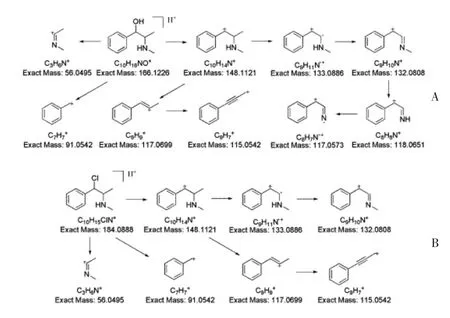
圖7 (偽)麻黃堿(A)和β-氯代甲基苯丙胺(B)CID碎裂途徑推測

圖6 (1S,2S)-β-氯代甲基苯丙胺/(1R,2S)-β-氯代甲基苯丙胺一級質譜圖(A)和二級質譜圖(B)
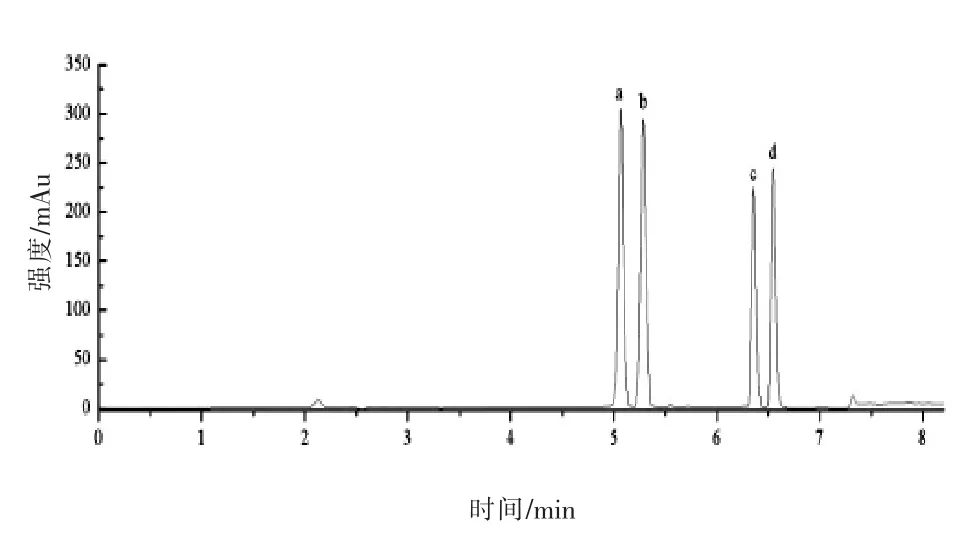
圖8 混合溶液LC-UV分析色譜圖
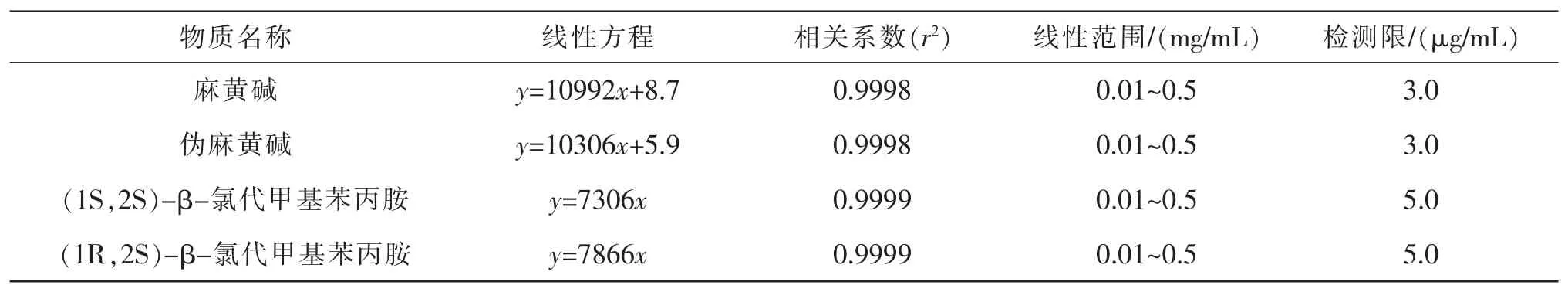
表3 4種化合物的線性范圍和檢出限
2.4 案例應用
案例樣品為福建非法制造β-氯代甲基苯丙胺案件中加工現場的繳獲物。LC-UV法分析結果為樣品中不含麻黃堿和偽麻黃堿,(1S,2S)-β-氯代甲基苯丙胺的純度(以C10H14ClN計,下同)為64.7%,(1S,2S)-β-氯代甲基苯丙胺的純度(以C10H14ClN計,下同)為8.2%,兩者的比例約為8∶1。進一步使用LC-MS分析發現,樣品中均有痕量麻黃堿和偽麻黃堿存在。判斷該加工廠已完成由麻黃堿/偽麻黃堿制β-氯代甲基苯丙胺的生產過程。
綜上,本方法使用液色譜質譜聯用法和液相色譜法對樣品中的麻黃堿、偽麻黃堿和(1S,2S)-β-氯代甲基苯丙胺、(1R,2S)-β-氯代甲基苯丙胺同時進行定性定量分析,方法簡便、靈敏。由于液相色譜的進樣和分離在常溫下進行,不會導致β-氯代甲基苯丙胺發生反應,同時可對光學異構體進行分離,對判斷加工廠的性質及其在制毒環節中的作用具有重要的意義。
∶
[1] Andrews KM.Ephedra’s Role as a Precursor in the Clandestine Manufacture of Methamphetamine[J].J Forensic Sci. 1995,(4):551-560.
[2] Allen AC,Kiser WO.Methamphetamine from Ephedrine. I.Chloroephedrines and Aziridines[J].J Forensic Sci,1987,(32):953-962.
[3] Barker WD,Antia U.A Study of the Ephedra in the Manufacture of Methamphetamine[J].Forensic Sci.Int,2007,166(2-3):102-109.
[4] Inoue T,Tanaka K,Ohmori T,et al.Impurity profiling Analysis of Methamphetamine Seized in Japan[J].Forensic Sci.Int,1994,(69):97-102.
[5] Cantrell TS,John B,Johnson L.et al.A Study of Impurities Found in Methamphetamine Synthesized from Ephedrine[J]. Forensic Sci.Int,1988,39(1):39-53.
[6] Barron RP,Kruegel AV,Moore JM.et al.Identification of Impurities in Illicit Methamphetamine Samples[J].J Assoc Off Anal.Chem,1974,57(5):1147-1158.
[7] Yoo YC,Lee WK,Chung HS.et al.Identification and Determination of Impurities in Illegal Drugs[J].Annu Rep. NISI,1985,(17):184-192.
[8] Ko BJ,Suh S,Suh YJ.et al.The Impurity Characteristics of Methamphetamine Synthesized by Emde and Nagai method[J].Forensic Sci.Int,2007,170(2-3):142-147.
[9] Salouros H,Collins M,George AV.et al.Isolation and Identification of Three By-Products Found in Methamphetamine Synthesized by the Emde Route[J].J Forensic Sci. 2010,55(3):605-615.
[10] 魏立青.對氯麻黃堿的綜合定性一例[D].第五次全國法醫學術交流會論文,1996:327-328.
[11] 李航麒,蘭暉,莊順,等.氯麻黃堿的檢驗[J].刑事技術,2015,40(6):477-479.
[12] Plotka JM,Morrison C,Adam D.et al.Chiral Analysis of Chloro Intermediates of Methylamphetamine by One-Dimensional and Multidimentional NMR and GC/MS[J]. Anal Chem,2012,(84):5625-5632.
(本文編輯:嚴慧)
Analysis of Ephedrine,Pseudophedrine,(1S,2S)-β-Chloro-methamphetamine and(1R,2S)-β-Chloro-methamphetamine
QIAN Zhen-hua1,LI Jing2,HUA Zhen-dong2
(1.Institute of Forensic Science,Ministry of Public Security,Beijing 100038,China;2.Drug Intelligence and Forensic Technology Center,Ministry of Public Security,Beijing 100193,China)
DF795.4
A
10.3969/j.issn.1671-2072.2017.05.006
1671-2072-(2017)05-0036-06
2017-02-17
中央級公益性科研院所基本科研業務費專項資金(2017JB030)
錢振華(1980—),女,助理研究員,主要從事毒品檢驗鑒定工作。E-mail:sunny1343@126.com。
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
