原發性家族性腦鈣化研究進展
2017-11-21 11:46:20陳悠岑志棟羅巍
中國現代神經疾病雜志 2017年7期
關鍵詞:基因突變
陳悠 岑志棟 羅巍
原發性家族性腦鈣化研究進展
陳悠 岑志棟 羅巍
原發性家族性腦鈣化是一組以雙側對稱性基底節區及其他腦區鈣化為影像學特點的神經變性病,可伴多種神經精神癥狀,具有高度臨床和遺傳異質性.目前已知的4種致病基因(SLC20A2、PDGFRB、PDGFB、XPR1)及其相關功能研究提示原發性家族性腦鈣化可能與細胞內外無機磷轉運障礙和血?腦屏障損害相關.本文擬對近年原發性家族性腦鈣化診斷標準、分子遺傳學機制、基因型與臨床表型相關性、治療等方面研究進展進行概述.
鈣質沉著癥; 腦疾病; 綜述
原發性家族性腦鈣化(PFBC)是一組以雙側對稱性基底節區鈣化,亦可累及小腦齒狀核、半卵圓中心、大腦皮質、中腦和腦橋等其他腦區為特征的神經系統遺傳性疾病,伴運動障礙、構音障礙、癲發作、共濟失調、認知功能障礙以及心境障礙、強迫癥等神經精神癥狀[1?2],具有高度臨床異質性和遺傳異質性,血清鈣、血清磷、甲狀旁腺激素(PTH)、堿性磷酸酶(ALP)等均于正常水平[3].本文擬對原發性家族性腦鈣化診斷標準、分子遺傳學機制、基因型與臨床表型相關性、治療等方面進展進行概述.
一、疾病命名
1850年,Delacour[4]報告首例顱內鈣化患者,臨床主要表現為雙下肢僵硬、無力伴震顫,尸檢顯示顱內鈣化灶以雙側紋狀體區為主.1930年,Fahr[5]報告1例癡呆患者,尸檢顯示半卵圓中心區和紋狀體區鈣化灶,盡管該個案報道既非首例,也未對理解疾病做出實質性貢獻,但"Fahr病(FD)"的稱法一直沿用至今,用以描述以基底節區為主的特發性顱內鈣化.此后文獻又陸續出現30余種命名,如Fahr綜合征(FS)、雙側紋狀體?蒼白球?齒狀核鈣質沉著(BSPDC)等[3].1977年,Boller等[6]提出"家族性特發性腦鈣化"的概念,首次強調疾病的家族遺傳性特點.自20世紀90年代開始,全世界范圍內廣泛開展關于疾病的遺傳學研究.早期遺傳學研究多以"特發性基底節區鈣化(IBGC)"命名[7],故在線人類孟德爾遺傳數據庫(OMIM)也沿用這一術語,根據報道時間順序,依次命名為IBGC1~6型.鑒于此類患者的顱內鈣化灶不僅局限于基底節區,且致病基因的發現使"特發性"名不副實,因此,近年來有學者建議將其命名為"原發性家族性腦鈣化"[8],本文也使用這一最新術語.
二、診斷標準
原發性家族性腦鈣化具有高度臨床異質性,可終身無癥狀,也可于不同年齡段發病,表現為嚴重程度不一、臨床癥狀各異的神經精神癥狀,因此,有學者認為臨床癥狀并非診斷必要條件[2].既往報道的致病基因明確的原發性家族性腦鈣化患者,臨床癥狀外顯率僅為61%(95%CI:0.560~0.720),而影像學外顯率達100%[9].作為診斷依據,影像學敏感性較高,但原發性家族性腦鈣化的影像學表現缺乏特異性,表現為雙側基底節區鈣化,伴或不伴其他腦區受累,這一特點也可見于正常老齡化過程或繼發于代謝性、自身免疫性、線粒體相關等50余種病因[10].因此,準確區分生理性或病理性鈣化以及排查其他可能病因對診斷至關重要.Nicolas等[2]于2013年率先提出一種評分體系,對CT顯示的顱內鈣化灶進行量化評價(表1),該方法對18個腦區進行評價,即左側和右側豆狀核、左側和右側尾狀核、左側和右側丘腦、左側和右側大腦皮質下白質、左側和右側內囊、大腦皮質、左側和右側小腦半球、小腦蚓部、左側和右側中腦、腦橋、延髓,再將各腦區評分相加獲得總鈣化分數(TCS).他們將600例神經科住院患者的頭部CT資料根據年齡分為3組(<40歲組、40~歲組和≥60歲組,每組各200例),計算年齡相關鈣化閾值,即該年齡段人群總鈣化分數第99百分位數值,結果顯示,3組患者年齡相關鈣化閾值分別為0、4和5分[2].Nicolas等[2]的診斷標準具備以下3項內容:(1)雙側豆狀核鈣化.(2)總鈣化分數大于相應年齡段年齡相關鈣化閾值.(3)排除其他可能引起基底節區鈣化的疾病,如特發性甲狀旁腺功能減退癥、假性甲狀旁腺功能減退癥、線粒體腦肌病(ME)、DiGeorge綜合征、Kenny?Caffey綜合征、Aicardi?Goutières綜合征、Coats疊加綜 合征、Labrune綜合征、Nasu?Hakola病、Cockayne綜合征、二氫蝶啶還原酶缺乏癥等[11].該診斷標準具有良好的循證醫學基礎,但針對的是法國人群,中國人群尚未開展此類研究,不同種族的生理性年齡相關鈣化閾值是否存有差異尚待進一步探究.
隨著致病基因的相繼鑒定與克隆,分子診斷技術對明確診斷原發性家族性腦鈣化也日趨重要.總結基因檢測陽性患者的臨床特點可能為制定基因檢測相關指南提供指導.2016年,Crütz等[12]提出,根據影像學和年齡預測致病性突變概率,并將腦區分為4個區域,即基底節區(包括尾狀核、殼核和蒼白球)、丘腦和下丘腦神經核團、小腦、大腦皮質,存在以下情況者提示基因檢測結果可能呈陽性:20~40歲患者上述4個區域中至少1個存在雙側鈣化灶,或者41~70歲患者上述4個區域中至少2個存在雙側鈣化灶.他們在1個原發性家族性腦鈣化家系(致病基因為SLC20A2基因)中采用這一方法,其靈敏度為100%、特異度為92.3%[12].該項診斷技術的準確性尚待在更多的原發性家族性腦鈣化家系或散發性患者中驗證.

表1 Nicolas等[2]的顱內鈣化評分體系Table 1. Intracranial Calcification Rating Scale(Nicolas,et al[2])
三、分子遺傳學機制
原發性家族性腦鈣化具有高度遺傳異質性,迄今共發現4種致病基因(表2).2012年,Wang等[17]采用全基因組微衛星標記連鎖分析,首次將原發性家族性腦鈣化致病基因定位于8p12~q11.23,并通過候選基因鑒定出首個致病基因SLC20A2.此后,其他研究團隊采用全外顯子測序(WES)或全基因組測序(WGS)先后鑒定出原發性家族性腦鈣化致病基因PDGFRB[18]、PDGFB[19]和XPR1[20].然而目前仍有大量散發性或有明確家族史的原發性家族性腦鈣化患者未攜帶已知基因的致病性突變,其致病基因尚待進一步鑒定和分析.
上述已知致病基因相關功能研究顯示,原發性家族性腦鈣化發病機制可能與細胞內外無機磷轉運障礙和血?腦屏障(BBB)損害相關.
1.SLC20A2和XPR1基因突變影響細胞內外無機磷轉運 (1)SLC20A2基因:SLC20A2基因可以編碼Ⅲ型鈉磷協同轉運體2(PiT2),后者高表達于神經元、星形膠質細胞和血管內皮細胞,主要分布于大腦皮質、基底節和黑質[21],與SLC20A1基因編碼的Ⅲ型鈉磷協同轉運體1(PiT1)共同調節細胞內外無機磷轉運過程.2012年,Wang等[17]在非洲爪蟾卵母細胞中導入原發性家族性腦鈣化的突變型PiT2蛋白(p.Ser601Trp、p.Ser601Leu、p.Thr595Met、p.Glu575Lys、p.Gly498Arg和p.Val42del)載體,結果顯示,突變型PiT2蛋白向細胞內轉運無機磷活性明顯下降,證實SLC20A2基因突變可以導致細胞內外無機磷轉運障礙.2013年,Jensen等[22]構建Slc20a2純合基因敲除小鼠模型,發現19周齡小鼠丘腦、基底節和大腦皮質等部位均出現鈣化灶,從動物模型水平證實PiT2蛋白功能缺失可以導致顱內鈣化;病理學檢查顯示,腦組織鈣鹽顆粒沿血管分布,與原發性家族性腦鈣化患者尸檢結果一致,提示突變型PiT2蛋白可能與血管鈣化有關.研究顯示,PiT2蛋白功能缺失可以引起局部細胞外無機磷水平升高,而細胞外高水平無機磷可以誘導血管平滑肌細胞鈣化[23?25].Wallingford等[26]通過小干擾RNA(siRNA)敲除小鼠血管平滑肌細胞SLC20A2基因,經高水平無機磷誘導后,實驗組鈣化程度顯著高于對照組,推測SLC20A2基因致病性突變可以導致PiT2蛋白功能缺失,局部無機磷代謝異常,使血管平滑肌細胞對高水平無機磷誘導鈣化的易感性增強,從而導致鈣化.(2)XPR1基因:XPR1基因編碼一種多重跨膜蛋白,表達于神經干細胞和腦組織,近年研究顯示其可以介導無機磷的輸出功能[27].2015年,Legati等[20]在人胚腎細胞293(HEK293)中導入原發性家族性腦鈣化突變型XPR1蛋白(p.Ser136Asn、 p.Leu140Pro、 p.Leu145Pro 和 p.Leu218Ser)載體,結果顯示,突變型XPR1蛋白介導的無機磷輸出功能損害,導致細胞內無機磷水平升高,細胞內鈣和無機磷沉積增加.
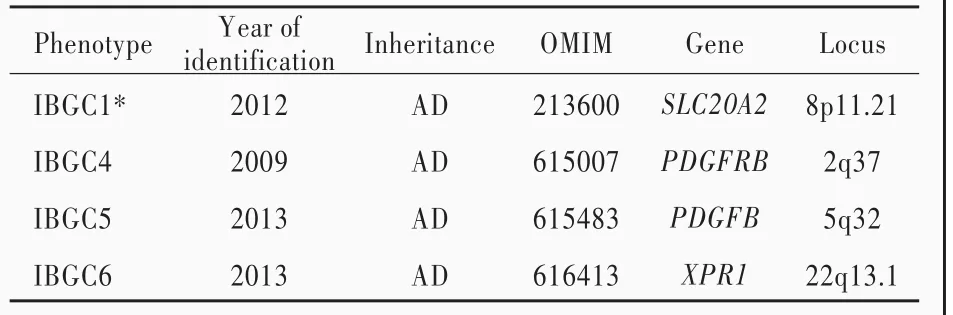
表2 原發性家族性腦鈣化致病基因Table 2. Causative genes of PFBC
2.PDGFRB和PDGFB基因突變破壞血?腦屏障完整性 (1)PDGFRB基因:PDGFRB基因編碼血小板源性生長因子受體β(PDGFRβ),屬Ⅲ型酪氨酸激酶受體家族,于神經元、脈絡叢、血管平滑肌細胞和周細胞中均有表達,主要分布于基底節和小腦齒狀核[28?30].PDGFRB基因功能獲得型突變與功能缺失型突變可以導致不同疾病,前者導致嬰兒型肌纖維瘤病[31]和Penttinen綜合征[32],后者導致原發性家族性腦鈣化[33].(2)PDGFB基因:PDGFB基因編碼血小板源性生長因子B(PDGFB),是PDGFRβ主要配體,表達于血管內皮細胞、巨核細胞和神經元[34].PDGFB/PDGFRβ信號轉導通路在腦血管周細胞發育過程中發揮關鍵作用,周細胞對維持血?腦屏障完整性至關重要[35].Keller等[19]構建PDGFB基因保留結構域純合敲除小鼠模型Pdgfbret/ret,結果顯示,小鼠中腦、丘腦、腦橋、基底前腦等部位均出現年齡依賴性鈣化灶.在PDGFB基因純合敲除小鼠模型Pdgfb-/-中導入2個拷貝的野生型PDGFB基因,可以防止顱內鈣化;而導入1個拷貝的野生型PDGFB基因仍可發生鈣化[19].PDGFRB基因與PDGFB基因的相關研究顯示,血?腦屏障完整性破壞可能是原發性家族性腦鈣化發病機制之一[18?19].
四、基因型與臨床表型的相關性
原發性家族性腦鈣化具有高度臨床異質性和遺傳異質性,探討基因型與臨床表型的相關性對診斷與治療的精準化具有重要意義.2013年,Nicolas等[2]在發現原發性家族性腦鈣化第2個致病基因PDGFRB后,首次對不同基因型與臨床表型的相關性進行研究,共納入25例攜帶SLC20A2基因(4例散發性患者,6例來自2個原發性家族性腦鈣化家系)或PDGFRB基因(2例散發性患者,13例來自1個原發性家族性腦鈣化家系)突變的原發性家族性腦鈣化患者,結果顯示,二者臨床癥狀無差異,但攜帶SLC20A2基因突變者總鈣化分數高于攜帶PDGFRB基因突變者.2015年,Tadic等[9]納入15篇文獻(2012年1月1日-2014年5月31日)共179例致病基因(SLC20A2、PDGFRB、PDGFB)明確的原發性家族性腦鈣化患者,其中162例來自25個原發性家族性腦鈣化家系,SLC20A2基因突變最為常見占66.5%(12例同時攜帶THAP1基因突變而未納入統計分析),PDGFB和PDGFRB基因突變分別占17.9%和8.9%,結果顯示,所有患者均存在顱內鈣化灶,病變部位依次為基底節區(70.6%)、皮質下白質(40.8%)、小腦(34.1%)和丘腦(28.5%);平均發病年齡(27.90±22.30)歲,攜帶不同致病基因的患者發病年齡差異未達到統計學意義;運動障礙是最常見的臨床癥狀,主要包括帕金森綜合征(12%)和肌張力障礙(19%).2017年,Batla等[36]納入20篇文獻(2012年1月1日-2016年11月7日)共137例致病基因(SLC20A2、PDGFRB、PDGFB、XPR1)明確的原發性家族性腦鈣化患者,來自34個原發性家族性腦鈣化家系,SLC20A2基因突變最為常見(54.74%,75/137),PDGFB、PDGFRB、XPR1基因突變分別占31.39%(43/137)、9.49%(13/137)和4.38%(6/137),結果顯示,與其他致病基因相比,攜帶SLC20A2基因突變的患者表現為帕金森病的概率更高,且丘腦和齒狀核受累更常見;攜帶PDGFB基因突變的患者伴頭痛的概率更高,但進一步研究顯示,在大多數家系中這一癥狀并未與疾病共分離,提示二者可能不存在因果關系;攜帶PDGFRB基因突變的患者發病年齡更早、頭痛和抑郁癥狀更常見,但差異未達到統計學意義;攜帶XPR1基因突變的患者出現認知功能障礙的概率更高、大腦皮質受累更常見,由于XPR1基因是新發現的致病基因且納入的病例數較少,攜帶該基因突變的患者臨床特點尚待進一步研究.上述研究均存在一定局限性,如選擇偏倚、某些致病基因的納入病例數較少、不同研究對臨床表型的描述無統一規范等.隨著相關術語的規范化,更多個案報道和人群篩查的研究,將有助于進一步明確基因型與臨床表型的相關性,為原發性家族性腦鈣化的診斷與治療提供指導.
五、治療
目前,原發性家族性腦鈣化主要以對癥治療為主,包括左旋多巴治療帕金森綜合征、抗癲藥物(AEDs)控制癲發作等,然而尚無有效方法可以阻斷或延緩顱內鈣化進展.Manyam[3]采用鈣拮抗劑尼莫地平進行治療,效果不甚理想.二膦酸鹽類藥由于其在骨骼重建相關疾病(如Paget病、骨質疏松癥、多發性骨髓瘤、惡性腫瘤骨轉移等)中的廣泛應用[37],獲得研究者們的青睞.二膦酸鹽類藥可以抑制鈣和無機磷沉積,阻斷非結晶型磷酸鈣轉變為羥基磷灰石,延緩磷灰石結晶進一步聚集[38],此外,二膦酸鹽類藥還可以透過血?腦屏障,這一特性使其成為絕佳的候選藥物.自1998年Loeb[39]率先采用二膦酸鹽類藥依替膦酸鈉治療1例原發性家族性腦鈣化患者后,陸續有研究者進行類似嘗試,發現部分患者某些臨床癥狀有所改善,但顱內鈣化程度并無變化[40?41].由于存在樣本量少、對照組缺乏、隨訪時間短等局限性,依替膦酸鈉的療效仍不確定,尚待進一步研究證實.近年來,隨著致病基因的相繼發現和相關功能研究的逐步深入,有助于闡明原發性家族性腦鈣化的分子遺傳學機制,可能為研發新的治療方法提供思路.
綜上所述,原發性家族性腦鈣化是一種具有高度臨床異質性和遺傳異質性的神經變性病.鑒別生理性鈣化與病理性鈣化是診斷原發性家族性腦鈣化的要點和難點.目前已鑒定和克隆出4種致病基因,即 SLC20A2、PDGFRB、PDGFB 和 XPR1,相關功能研究提示其發病機制可能與細胞內外無機磷轉運障礙和血?腦屏障損害相關.由于研究納入的樣本量較少、選擇偏倚、描述用語不規范等局限性,原發性家族性腦鈣化基因型與臨床表型的相關性尚不確定,尚待進一步研究.目前主要以對癥治療為主.未來隨著更多致病基因的鑒定和相關功能研究的開展,對原發性家族性腦鈣化將有更深入的認識,有助于進一步規范診斷流程、闡明分子遺傳學機制、明確基因型與臨床表型的相關性、研發有效的治療方法等.
[1]Zhao YC.Advances in idiopathic basal ganglia calcification.Zhongguo Xian Dai Shen Jing Ji Bing Za Zhi,2011,11:27?29.[趙迎春.原發性基底節鈣化研究進展.中國現代神經疾病雜志,2011,11:27?29.]
[2]Nicolas G,Pottier C,Charbonnier C,Guyant?Maréchal L,Le Ber I,Pariente J,Labauge P,Ayrignac X,Defebvre L,Maltête D, Martinaud O,Lefaucheur R, Guillin O, Wallon D,Chaumette B,Rondepierre P,Derache N, Fromager G,Schaeffer S,Krystkowiak P,Verny C,Jurici S,Sauvée M,Vérin M,Lebouvier T,Rouaud O,Thauvin?Robinet C,Rousseau S,Rovelet?Lecrux A,Frebourg T,Campion D,Hannequin D;French IBGC Study Group.Phenotypic spectrum of probable and genetically?confirmed idiopathic basal ganglia calcification.Brain,2013,136:3395?3407.
[3]Manyam BV.Whatis and whatis not'Fahr's disease'.Parkinsonism Relat Disord,2005,11:73?80.
[4]Delacour A.Ossification des capillaires du cerveau.Ann Med Psychol,1850,2:458?461.
[5]Fahr T.Idiopathische verkalkung der hirngef?sse.Zbl Allg Pathol Pathol Anat,1930,50:129?133.
[6]BollerF,BollerM,GilbertJ.Familialidiopathic cerebral calcifications.J Neurol Neurosurg Psychiatry,1977,40:280?285.
[7]Kuroiwa Y,Boller F,Boller M.Computed tomographic demonstration of idiopathic familial basal ganglia calcification.Comput Radiol,1983,7:141?143.
[8]Lemos RR,Ferreira JB,Keasey MP,Oliveira JR.An update on primary familial brain calcification.Int Rev Neurobiol,2013,110:349?371.
[9]Tadic V,Westenberger A,Domingo A,Alvarez?Fischer D,Klein C,Kasten M.Primary familial brain calcification with known gene mutations:a systematic review and challenges of phenotypic characterization.JAMA Neurol,2015,72:460?467.
[10]Baba Y,Broderick DF,Uitti RJ,Hutton ML,Wszolek ZK.Heredofamilialbrain calcinosis syndrome.Mayo Clin Proc,2005,80:641?651.
[11]Deng H,Zheng W,Jankovic J.Genetics and molecular biology of brain calcification.Ageing Res Rev,2015,22:20?38.
[12]Grütz K,Volpato CB,Domingo A,Alvarez?Fischer D,Gebert U,Schifferle G,Buffone E,Wszolek ZK,Rademakers R,Ferbert A,HicksAA,Klein C,PramstallerPP,WestenbergerA.Primary familial brain calcification in the'IBGC2'kindred:all linkage roads lead to SLC20A2.Mov Disord,2016,31:1901?1904.
[13]Dai X,Gao Y,Xu Z,Cui X,Liu J,Li Y,Xu H,Liu M,Wang QK,Liu JY.Identification ofa novelgenetic locus on chromosome 8p21.1-q11.23 for idiopathic basal ganglia calcification.Am J Med Genet B Neuropsychiatr Genet,2010,153B:1305?1310.
[14]Geschwind DH,Loginov M,Stern JM.Identification of a locus on chromosome 14q for idiopathic basal ganglia calcification(Fahr disease).Am J Hum Genet,1999,65:764?772.
[15]Volpato CB,De Grandi A,Buffone E,Facheris M,Gebert U,Schifferle G,Sch?nhuber R,Hicks A,Pramstaller PP.2q37 as a susceptibility locus for idiopathic basal ganglia calcification(IBGC)in a large South Tyrolean family.J Mol Neurosci,2009,39:346?353.
[16]Hsu SC,Sears RL,Lemos RR,Quintáns B,Huang A,Spiteri E,Nevarez L,Mamah C,Zatz M,Pierce KD,Fullerton JM,Adair JC,Berner JE,Bower M,Brodaty H,Carmona O,Dobricic V,Fogel BL,García?Estevez D,Goldman J,Goudreau JL,Hopfer S,Jankovic M,Jaumà S,Jen JC,Kirdlarp S,Klepper J,Kostic V,LangAE,LinglartA,MaisenbacherMK,Manyam BV,MazzoniP,Miedzybrodzka Z,Mitarnun W,MitchellPB,Mueller J,Novakovic I,Paucar M,Paulson H,Simpson SA,Svenningsson P,Tuite P,Vitek J,WetchaphanphesatS,Williams C,Yang M,Schofield PR,de Oliveira JR,Sobrido MJ,Geschwind DH,Coppola G.Mutations in SLC20A2 are a major cause of familial idiopathic basal ganglia calcification.Neurogenetics,2013,14:11?22.
[17]Wang C,Li Y,Shi L,Ren J,Patti M,Wang T,de Oliveira JR,Sobrido MJ,Quintáns B,Baquero M,Cui X,Zhang XY,Wang L,Xu H,Wang J,Yao J,Dai X,Liu J,Zhang L,Ma H,Gao Y,Ma X,Feng S,Liu M,Wang QK,Forster IC,Zhang X,Liu JY.Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis.Nat Genet,2012,44:254?256.
[18]Nicolas G,Pottier C,Maltête D,Coutant S,Rovelet?Lecrux A,Legallic S,Rousseau S,Vaschalde Y,Guyant?Maréchal L,Augustin J,Martinaud O,Defebvre L,Krystkowiak P,Pariente J,Clanet M,Labauge P,Ayrignac X,Lefaucheur R,Le Ber I,Frébourg T,Hannequin D,Campion D.Mutation ofthe PDGFRB gene as a cause of idiopathic basal ganglia calcification.Neurology,2013,80:181?187.
[19]Keller A,Westenberger A,Sobrido MJ,García ?Murias M,Domingo A,Sears RL,Lemos RR,Ordo?ez?Ugalde A,Nicolas G,da Cunha JE,Rushing EJ,Hugelshofer M,Wurnig MC,Kaech A,Reimann R,Lohmann K,Dobricic V,Carracedo A,Petrovic I,Miyasaki JM,Abakumova I,M?e MA,Raschperger E,Zatz M,Zschiedrich K,Klepper J,Spiteri E,Prieto JM,Navas I,Preuss M,Dering C,Jankovic M,Paucar M,Svenningsson P,Saliminejad K,Khorshid HR,Novakovic I,Aguzzi A,Boss A,Le Ber I,Defer G,Hannequin D,Kostic VS,Campion D,Geschwind DH,Coppola G,Betsholtz C,Klein C,Oliveira JR.Mutations in the gene encoding PDGF?B cause brain calcifications in humans and mice.Nat Genet,2013,45:1077?1782.
[20]Legati A,Giovannini D,Nicolas G,López?Sánchez U,Quintáns B,Oliveira JR,Sears RL,Ramos EM,Spiteri E,Sobrido MJ,Carracedo á,Castro ?Fernández C,Cubizolle S,Fogel BL,Goizet C,Jen JC,Kirdlarp S,Lang AE,Miedzybrodzka Z,Mitarnun W,Paucar M,Paulson H,Pariente J,Richard AC,Salins NS,Simpson SA,Striano P,Svenningsson P,Tison F,Unni VK,Vanakker O,Wessels MW,Wetchaphanphesat S,YangM,BollerF,Campion D,Hannequin D,Sitbon M,Geschwind DH,Battini JL,Coppola G.Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export.Nat Genet,2015,47:579?581.
[21]Lagrue E,Abe H,Lavanya M,Touhami J,Bodard S,Chalon S,Battini JL,Sitbon M,Castelnau P.Regional characterization of energy metabolism in the brain of normal and MPTP?intoxicated mice using new markers of glucose and phosphate transport.J Biomed Sci,2010,17:91.
[22]Jensen N,Schr?derHD,Hejb?lEK,FüchtbauerEM,de Oliveira JR,Pedersen L.Loss of function of Slc20a2 associated with familial idiopathic basal ganglia calcification in humans causes brain calcifications in mice.J Mol Neurosci,2013,51:994?999.
[23]Giachelli CM,Jono S,Shioi A,Nishizawa Y,Mori K,Morii H.Vascular calcification and inorganic phosphate.Am J Kidney Dis,2001,38:S34?37.
[24]Jono S,McKee MD,Murry CE,Shioi A,Nishizawa Y,Mori K,Morii H,Giachelli CM.Phosphate regulation of vascular smooth muscle cell calcification.Circ Res,2000,87:E10?17.
[25]Lomashvili KA,Cobbs S,Hennigar RA,Hardcastle KI,O'Neill WC. Phosphate?induced vascular calcification: role of pyrophosphate and osteopontin.J Am Soc Nephrol,2004,15:1392?1401.
[26]Wallingford MC,Chia JJ,Leaf EM,Borgeia S,Chavkin NW,Sawangmake C,Marro K,Cox TC,Speer MY,Giachelli CM.SLC20A2 deficiency in mice leads to elevated phosphate levels in cerbrospinalfluid and glymphatic pathway?associated arteriolar calcification,and recapitulates human idiopathic basal ganglia calcification.Brain Pathol,2017,27:64?76.
[27]Giovannini D,Touhami J,Charnet P,Sitbon M,Battini JL.Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans.Cell Rep,2013,3:1866?1873.
[28]Hutchins JB,Jefferson VE.Developmentaldistribution of platelet?derived growth factor in the mouse central nervous system.Brain Res Dev Brain Res,1992,67:121?135.
[29]Ishii Y,Oya T,Zheng L,Gao Z,Kawaguchi M,Sabit H,Matsushima T,Tokunaga A,Ishizawa S,Hori E,Nabeshima Y,Sasaoka T,Fujimori T,Mori H,Sasahara M.Mouse brains deficient in neuronal PDGF receptor?beta develop normally but are vulnerable to injury.J Neurochem,2006,98:588?600.
[30]Lindahl P,Johansson BR,Levéen P,Betsholtz C.Pericyte loss and microaneurysm formation in PDGF?B?deficient mice.Science,1997,277:242?245.
[31]Cheung YH,Gayden T,Campeau PM,LeDuc CA,Russo D,Nguyen VH,Guo J,Qi M,Guan Y,Albrecht S,Moroz B,Eldin KW,Lu JT,Schwartzentruber J,Malkin D,Berghuis AM,Emil S,Gibbs RA,Burk DL,Vanstone M,Lee BH,Orchard D,Boycott KM,ChungWK,JabadoN.A recurrent PDGFRB mutation causes familial infantile myofibromatosis.Am J Hum Genet,2013,92:996?1000.
[32]Johnston JJ,Sanchez?Contreras MY,Keppler?Noreuil KM,Sapp J,Crenshaw M,Finch NA,Cormier?Daire V,Rademakers R,Sybert VP,Biesecker LG.A point mutation in PDGFRB causes autosomal?dominant penttinen syndrome.Am J Hum Genet,2015,97:465?474.
[33]Sanchez?Contreras M,Baker MC,Finch NA,Nicholson A,Wojtas A,Wszolek ZK,Ross OA,Dickson DW,Rademakers R.Genetic screening and functional characterization of PDGFRB mutations associated with basal ganglia calcification of unknown etiology.Hum Mutat,2014,35:964?971.
[34]Andrae J,Gallini R,Betsholtz C.Role of platelet?derived growth factors in physiology and medicine.Genes Dev,2008,22:1276?1312.
[35]Daneman R,Zhou L,Kebede AA,Barres BA.Pericytes are required for blood?brain barrier integrity during embryogenesis.Nature,2010,468:562?566.
[36]Batla A,Tai XY,Schottlaender L,Erro R,Balint B,Bhatia KP.Deconstructing Fahr's disease/syndrome of brain calcification in the era of new genes.Parkinsonism Relat Disord,2017,37:1?10.
[37]Drake MT,Clarke BL,Khosla S.Bisphosphonates:mechanism of action and role in clinical practice.Mayo Clin Proc,2008,83:1032?1045.
[38]Fleisch H.Bisphosphonates:a new class of drugs in diseases of bone and calcium metabolism.Recent Results Cancer Res,1989,116:1?28.
[39]Loeb JA.Functional improvement in a patient with cerebral calcinosis using a bisphosphonate.Mov Disord,1998,13:345?349.
[40]Loeb JA,Sohrab SA,Huq M,Fuerst DR.Brain calcifications induce neurological dysfunction that can be reversed by a bone drug.J Neurol Sci,2006,243:77?81.
[41]Oliveira JR,Oliveira MF.Primary brain calcification in patients undergoing treatment with the biphosphanate alendronate.Sci Rep,2016,6:22961.
Recent study on primary familial brain calcification
CHEN You,CEN Zhi?dong,LUO Wei
Department of Neurology,the Second Affiliated Hospital of Zhejiang University School of Medcine,Hangzhou 310009,Zhejiang,China
LUO Wei(Email:luoweirock@126.com)
Primary familialbrain calcification (PFBC),characterized by bilateral,symmetric calcifications in basal ganglia and other brain regions and visualized in neuroimaging and neuropsychiatric manifestations variable in type and severity,is a neurodegenerative disorder with clinical and genetic heterogeneity.The discovery of causative genes(namely SLC20A2,PDGFRB,PDGFB and XPR1)and functional studies indicated that PFBC may be related to inorganic phosphate transport dysfunction and blood?brain barrier deficiency.Since the understanding of PFBC has advanced dramatically in recent years,this review focuses on diagnosis,molecular genetics,genotype?phenotype relationship and treatment in PFBC.
Calcinosis; Brain diseases; Review
This study was supported by the National Natural Science Foundation of China(No.81571089).
10.3969/j.issn.1672?6731.2017.07.003
國家自然科學基金資助項目(項目編號:81571089)
310009杭州,浙江大學醫學院附屬第二醫院神經內科
羅巍(Email:luoweirock@126.com)
2017?06?05)
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
