頭部震顫伴小腦萎縮一例臨床表型及基因突變分析
2017-11-21 11:46:23謝坤銘顧衛紅郝瑩陳園園張瑾張鑫
中國現代神經疾病雜志 2017年7期
謝坤銘 顧衛紅 郝瑩 陳園園 張瑾 張鑫
頭部震顫伴小腦萎縮一例臨床表型及基因突變分析
謝坤銘 顧衛紅 郝瑩 陳園園 張瑾 張鑫
目的通過對1例頭部震顫伴小腦萎縮患者臨床表型和基因檢測結果進行綜合分析,明確診斷疾病并探討基因檢測結果的解讀方法.方法與結果采集1例30歲男性患者臨床表型,進行二代基因測序和Sanger測序驗證,通過中文人類表型標準用語、基因檢索工具Phenomizer、Ensembl數據庫、在線人類孟德爾遺傳數據庫相關信息,對基因檢測結果進行解讀.結果顯示,患者存在脊髓小腦共濟失調19型(SCA19型)致病基因KCND3基因雜合突變c.1057A>G(p.Ser353Gly),其父母均未攜帶該突變基因;患者還存在帕金森病20型致病基因SYNJ1基因雜合突變c.4436C>T(p.Thr1479Ile),其母攜帶該突變基因.表型相似度分析顯示,患者表型與SCA19型一致,KCND3基因變異位點c.1057A>G在不同物種同源基因中具有高度保守性.結論通過對患者臨床表型和基因檢測結果綜合分析,KCND3基因雜合突變c.1057A>G(p.Ser353Gly)為致病性突變.
脊髓小腦共濟失調; 表型; 基因; 突變
共濟失調作為常見的神經系統疾病表現,涉及 多種病因,包括單基因遺傳病、散發性神經變性病和獲得性疾病.從遺傳學角度看,神經系統遺傳性疾病約占所有遺傳性疾病的60%,遺傳性共濟失調占神經系統遺傳性疾病的10%~15%,迄今已報道100余種類型[1].遺傳模式可以分為常染色體顯性遺傳(AD)、常染色體隱性遺傳(AR)、X連鎖遺傳和線粒體母系遺傳,具有高度臨床異質性和遺傳異質性,其中,脊髓小腦共濟失調(SCA)呈常染色體顯性遺傳[2?3],臨床主要表現為平衡障礙、肢體協調障礙、步態不穩、構音障礙和眼球運動障礙等,且伴復雜神經系統損害,如錐體系、錐體外系、視覺、聽覺、脊髓和周圍神經系統損害等,主要病理損害部位為小腦、腦干和脊髓.目前報道的亞型達40余種[4?5].本文詳細分析1例頭部震顫伴小腦萎縮患者的臨床表型并對其基因檢測結果進行深入解讀.
病例資料
一、臨床特征
患者 男性,30歲,因頭頸部不自主晃動2年,于2013年11月6日至中日友好醫院運動障礙與神經遺傳病專病門診就診.患者2年前無明顯誘因出現頭頸部不自主晃動,發作時間持續1~2 min或數天不等,肩部晃動僵硬,緊張和精細動作時易發作,進行性加重;言語模糊,構音不清,偶有吞咽困難,睡眠時好時壞,可見胸悶、呼吸困難、反復發作的神經性皮炎.既往史和個人史無特殊.父母身體健康,非近親婚配,無明確家族性遺傳性疾病病史.門診神經系統檢查:神志清楚,輕度構音障礙;眼球各向活動不充分,外展露白,未見眼震;頭部陣發性晃動;四肢肌力5級,雙上肢肌張力降低,腱反射減弱,雙下肢肌張力正常,腱反射正常,足跖反射正常,病理反射未引出;雙側指鼻試驗、跟?膝?脛試驗穩準.簡易智能狀態檢查量表(MMSE)評分正常.實驗室檢查各項指標均于正常值范圍.影像學檢查:頭部MRI顯示小腦重度萎縮,腦干中度萎縮(圖1).神經電生理學檢查:心電圖、肌電圖和腦電圖均未見明顯異常.結合臨床表現和家族史,臨床擬診為常染色體隱性遺傳性共濟失調.予金剛烷胺50 mg/次、2次/d,苯海索(安坦)1 mg/次、2次/d,氯硝西泮1 mg/d口服治療.至2016年3月復診時頭部晃動明顯緩解.
二、基因檢測
分別抽取患者及其父母外周靜脈血5 ml,送檢北京金準基因科技有限責任公司,進行3994種人類孟德爾遺傳性疾病致病基因外顯子二代基因測序和家系驗證.對候選變異位點采用Sanger測序進行家系驗證,提示患者存在2種可能的致病性突變:(1)KCND3基因雜合突變c.1057A>G(p.Ser353Gly).Sanger測序顯示其父母均未攜帶該突變基因(圖2).KCND3基因與SCA19型相關,呈常染色體顯性遺傳,理論上1條染色體突變即可致病.(2)SYNJ1基因雜合突變c.4436C>T(p.Thr1479Ile).Sanger測序證實此突變來自其母(圖3).SYNJ1基因與帕金森病20型(PARK20型)相關,呈常染色體隱性遺傳,理論上必須2條等位染色體同時出現致病性突變方可致病(即純合突變或復合雜合突變致病).
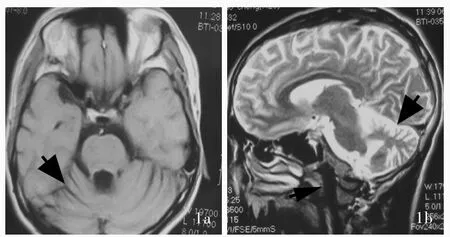
圖1 頭部MRI檢查顯示,小腦呈現重度萎縮(粗箭頭所示),腦干呈現中度萎縮(細箭頭所示)1a 橫斷面T1WI 1b 矢狀位T2WIFigure1 Head MRIshowed severecerebellaratrophy(thick arrows indicate)and moderate brain stem atrophy(thin arrow indicates). AxialT1WI(Panel1a). SagittalT2WI(Panel 1b).
三、突變的致病性分析
突變的致病性分析步驟為:(1)將核心表型轉化為中文人類表型標準用語/人類表型標準用語(CHPO/HPO,http://www.chinahpo.org/).(2)候選基因篩選,根據對應的CHPO/HPO詞匯,采用基因檢索工具Phenomizer(http://compbio.charite.de/phenomizer/)檢索候選基因.(3)根據美國醫學遺傳學和基因組學會(ACMG)制定的變異位點分級指南(http://wintervar.wglab.org/),輸入突變基因信息,結合核心表型、家族史和遺傳模式,即可判斷突變基因的致病性."Pathogenic"表示已知致病性突變,"likely pathogenic"表示疑似致病性突變,"VUS"表示臨床意義不明的突變,"likely benign"表示疑似良性突變,"benign"表示良性突變.(4)保守性分析,通過 Ensembl數據庫(http://useast.ensembl.org/index.html)獲取多物種相關基因序列,采用BioEdit軟件(http://www.bioedit.com/)比對變異位點在不同物種的序列保守性.(5)表型相似度分析,通過檢索在線人類孟德爾遺傳數據庫(http://www.omim.org/)比對疾病表型與患者表型.
1.核心表型轉化為中文人類表型標準用語 采用CHPO搜索引擎檢索本文患者的核心表型,結果參見表1.
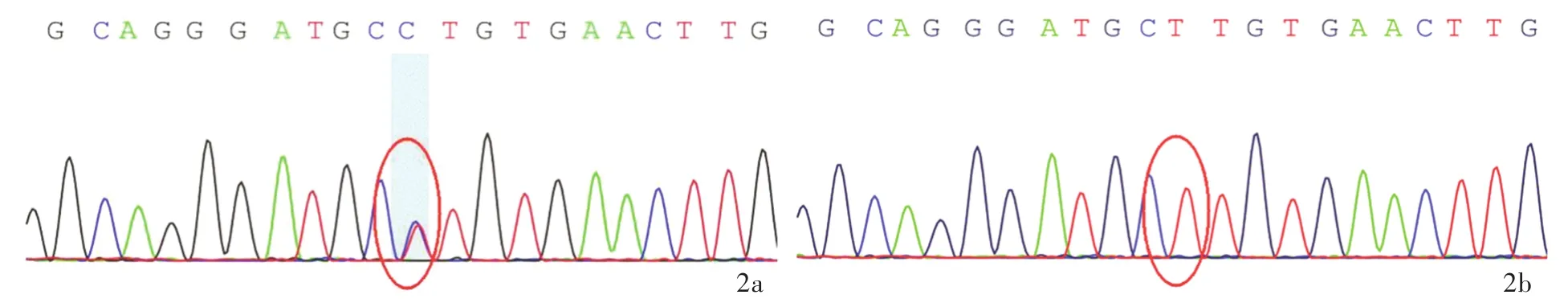
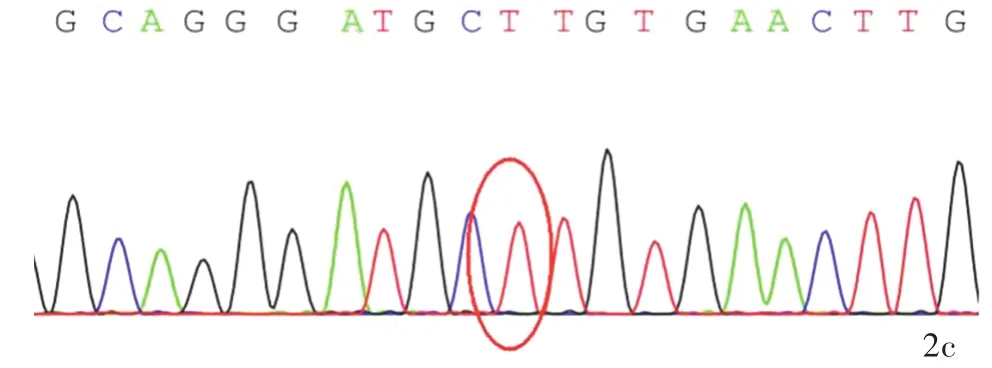
圖2 該例患者及其父母KCND3基因Sanger測序所見 2a 患者存在KCND3基因c.1057A>G(p.Ser353Gly)雜合突變(紅色圓圈所示)2b 其父不攜帶KCND3基因突變(紅色圓圈所示)2c其母不攜帶KCND3基因突變(紅色圓圈所示)Figure 2 The Sanger sequencing of KCND3 gene of the patient and his parents.Heterozygous mutation of KCND3 gene c.1057A>G(p.Ser353Gly)in the patient(red circle indicates,Panel 2a).No KCND3 gene mutation in the patient's father(red circle indicates,Panel 2b)and mother(red circle indicates,Panel 2c).
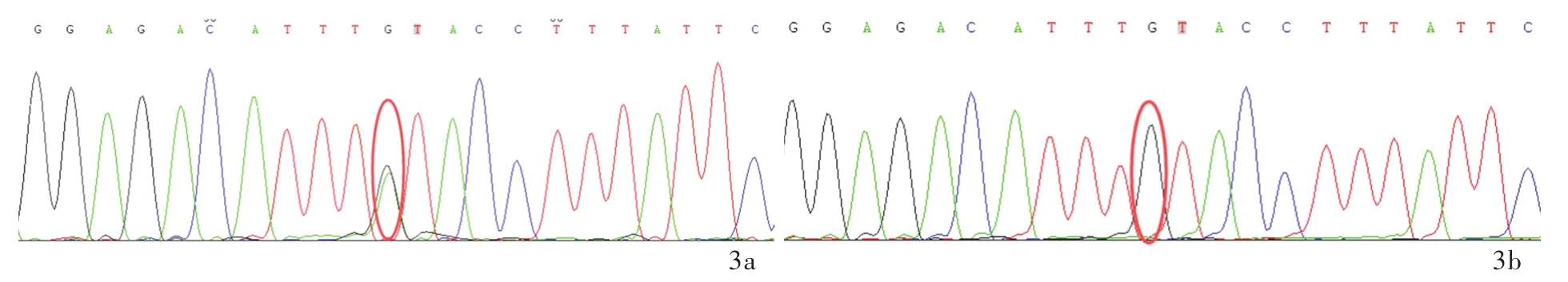
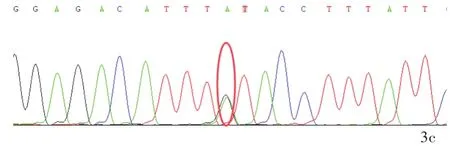
圖3 該例患者及其父母SYNJ1基因Sanger測序所見 3a 患者存在SYNJ1基因c.4436C>T(p.Thr1479Ile)雜合突變(紅色圓圈所示) 3b 其父不攜帶SYNJ1基因突變(紅色圓圈所示)3c 其母存在SYNJ1基因c.4436C>T(p.Thr1479Ile)雜合突變(紅色圓圈所示)Figure 3 The Sanger sequencing of SYNJ1 gene of the patient and his parents.Heterozygous mutation of SYNJ1 gene c.4436C>T(p.Thr1479Ile)in the patient(red circle indicates,Panel 3a).No SYNJ1 gene mutation in the patient's father(red circle indicates,Panel 3b).Heterozygous mutation of SYNJ1 gene c.4436C>T(p.Thr1479Ile)in the patient's mother(red circle indicates,Panel 3c).
2.基于人類表型標準用語篩選候選基因 通過基因檢索工具Phenomizer,篩選致病基因,結果顯示,相關度最大的基因為KCND3基因(P=0.000).
3.確定致病性突變 根據美國醫學遺傳學和基因組學會制定的變異位點分級指南,KCND3基因為"pathogenic",即致病性突變;SYNJ1基因為"likely pathogenic",即疑似致病性突變.
4.保守性分析 通過Ensemble數據庫檢索KCND3基因及其他物種同源基因序列,采用BioEdit軟件進行比對分析,結果顯示,變異位點在不同物種的進化中具有高度保守性(圖4).
5.表型相似度分析 通過在線人類孟德爾遺傳數據庫檢索KCND3和SYNJ1基因相關疾病表型,與患者表型進行比對.核心表型為小腦萎縮、頭頸部不自主晃動、腱反射減弱、構音障礙和吞咽困難,與KCND3基因的疾病表型(SCA19型)高度相似,提示為致病基因(表2).SYNJ1基因的疾病表型為PARK20型,表現為面具臉、目光凝視、眼瞼失用癥、掃視障礙、核上性凝視麻痹、下頜震顫、動作遲緩、肌僵直、拖步、姿勢不穩、震顫、肌張力障礙、認知功能障礙和早發型癲發作,頭部MRI顯示大腦皮質萎縮[6?7],與患者表型不符,排除SYNJ1基因致病的可能.經基因檢測最終明確診斷為SCA19型,繼續予藥物治療以緩解相應癥狀.
討 論
脊髓小腦共濟失調呈常染色體顯性遺傳,自1993年SCA1型致病基因定位以來,根據研究者對其致病基因定位的時間順序,由國際人類基因組組織基因命名委員會(HGNC)命名,至2017年5月已定位43個致病基因位點,其中28種致病基因已被克隆(http://neuromuscular.wustl.edu/ataxia/domatax.html).脊髓小腦共濟失調家系中大部分為致病基因動態突變,采用毛細管電泳片段分析法可檢出.隨著二代基因測序技術的發展,越來越多的非動態突變逐漸被發現,其致病性尚待進一步明確.

表1 該例患者核心表型及其對應的CHPO/HPOTable 1. The core phenotype and corresponding CHPO/HPO of the patient

圖4 保守性分析顯示,KCND3基因變異位點c.1057A>G在不同物種同源基因中具有高度保守性Figure 4 Conservation analysis showed the variation locus of KCND3 gene c.1057A>G was highly conservative with the homologous gene in different species.
該例患者28歲起病,無明確家族史,最初考慮為常染色體隱性遺傳性共濟失調,通過二代基因測序并結合臨床表型分析,檢出SCA19型相關基因KCND3基因雜合突變c.1057A>G(p.Ser353Gly),為新發突變,且在進化過程中高度保守.
SCA19型發病年齡多樣化,自青年至中年均可發病,進展緩慢.已報道的家系多有小腦萎縮癥狀,以頭部晃動發病,伴輕至中度構音障礙,其他癥狀還包括凝視誘發眼震,吞咽困難,小腦、步態、軀干、肢體共濟失調,腱反射減弱,姿勢性震顫,肌陣攣,認知功能障礙和踝振動覺受損等[8].SCA19型最早發現于一荷蘭家系,先證者約31歲時以頭部晃動發病,繼而出現構音障礙,逐漸出現行走困難,神經系統查體顯示不規則震顫,頻率緩慢,姿勢變化和站立時頭頸部不自主晃動,細小眼震,眼球各向活動充分,關節位置覺受損、雙足振動覺減退,四肢協調性受到嚴重影響,表現出辨距不良和快復輪替動作笨拙,共濟失調步態[9].Verbeek等[10]通過全基因組測序(WGS)將致病基因定位于1p21~q21.2003年一中國脊髓小腦共濟失調家系致病基因定位于1p21~q23,當時被國際人類基因組組織基因命名委員會命名為SCA22型[11].但在2004年,Schelhaas等[12]發現SCA19型和SCA22型的基因突變位點相重合,至此認為二者為脊髓小腦共濟失調的同一亞型.2012年,Duarri等[13]的研究顯示,KCND3基因為SCA19型致病基因.
KCND3蛋白系編碼電壓門控性鉀離子通道(VGKC)的Shal家族α亞單位,對細胞膜復極化具有重要作用[14],包括6個跨膜片段和細胞內氨基末端(N末端)和羧基末端(C末端).逆轉錄?聚合酶鏈反應(RT?PCR)和測序分析證實1個剪切位點突變KCND3L,插入編碼1個含19個氨基酸的多肽鏈,包含1個磷酸化位點,其中較短亞型為KCND3S[15].Northern blotting法檢測顯示,長度為8.50X103bp的轉錄本在腦和心臟呈高表達,而在腎臟、肝臟、肺、胰腺、脾或骨骼肌中未檢測到[16].這種長轉錄本主要見于丘腦、尾狀核、白質和骨骺,短轉錄本主要見于額葉、枕葉和小腦皮質[17].1999年首次在人類大腦和心臟中克隆出KCND3基因,RT?PCR法發現僅KCND3基因長片段在心臟表達[18],包含7個外顯子,長度約25X 103bp,而較短亞型由6個外顯子編碼.KCND3基因突變導致其編碼的電壓門控性鉀離子通道特性改變,引起智力障礙、癲發作、注意力缺陷、多動、斜視和口唇失用等癥狀[19].
該例患者頭頸部不自主晃動,肩部晃動僵硬,緊張和精細動作時易發作,進行性加重,言語模糊;神經系統檢查,輕度構音障礙,眼球各向活動不充分,未見眼震,陣發性頭部晃動,四肢肌力5級,雙上肢肌張力偏低,腱反射減弱,雙下肢肌張力正常,腱反射適中,足跖反射正常,雙側指鼻試驗、跟?膝?脛試驗穩準;頭部MRI顯示明顯小腦萎縮,上述表型與荷蘭SCA19型家系臨床表型相似.
SYNJ1基因相關疾病表型為PARK20型,臨床特點為運動遲緩、肌肉僵直、靜止性震顫、姿勢不穩、核上性凝視麻痹、癡呆、肌張力障礙和全面性強直?陣攣發作(GTCS)[20],左旋多巴或其他擬多巴胺類藥治療有效[21].該例患者帕金森病癥狀并不明顯,且該突變位點源自其母,其父無突變,該基因為常染色體隱性遺傳,因此考慮SYNJ1基因致病的可能性不大.
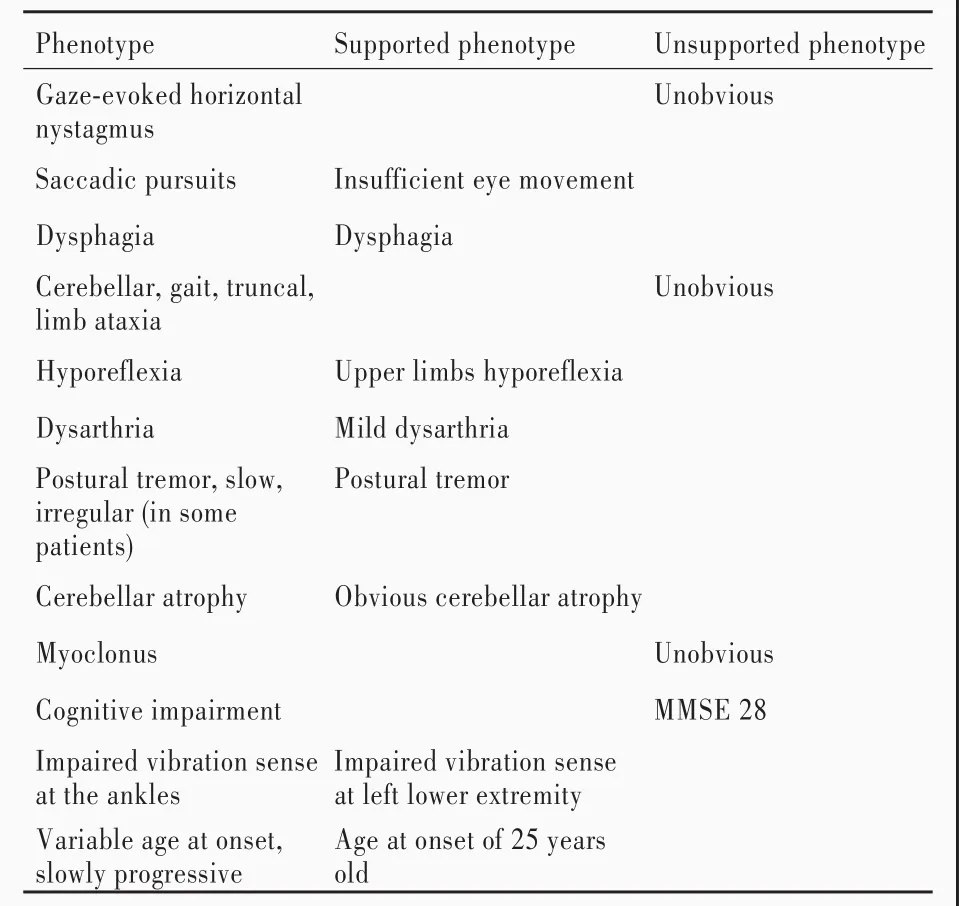
表2 該例患者KCND3基因相關疾病表型與患者表型相似度分析Table 2. Phenotypic similarity analysis of the patient
在對該例患者臨床表型和基因檢測結果進行綜合分析解讀過程中,我們采用CHPO/HPO,有助于表型標準化并連接基因數據庫進行解讀.HPO旨在提供人類疾病中用于描述表型異常的標準詞匯,從醫學文獻、Orphanet數據庫、DECIPHER數據庫和在線人類孟德爾遺傳數據庫獲得的信息進行開發,目前包含11 896個名詞,同時還提供一套針對約4000種疾病的注釋.HPO一直持續進行詞條維護和完善.目前國內大部分醫療和科研工作者所使用的表型描述詞匯并無統一標準,大多數醫院信息系統中記錄的臨床表型數據均為非結構化的自然詞匯,給人類疾病尤其是遺傳性疾病的臨床研究和交流帶來不便,對后期數據挖掘和分析也造成很大困擾,因此,在中國建立一套標準的臨床表型詞匯非常重要.CHPO作為開放平臺,旨在聯合各相關領域專業人士,逐步建立中文臨床表型詞匯標準,并指導和服務于中文使用者的臨床和科研工作,目前已建立搜索引擎http://www.chinahpo.org/.
本研究通過對1例無明確家族史的頭部震顫伴小腦萎縮患者的臨床表型和基因數據進行綜合分析,探討表型標準化結合分析軟件工具進行基因檢測選點和結果解讀的方法和流程:(1)將患者核心表型轉化為CHPO/HPO.(2)將患者的HPO結果輸入基因檢索工具,如Phenomizer,篩選可能的候選致病基因.(3)將二代基因測序檢出的基因變異位點與上述分析結果相結合進行選點.(4)根據美國醫學遺傳學和基因組學會制定的變異位點分級指南進行致病性評價.(5)表型相似度分析,基于在線人類孟德爾遺傳數據庫記錄的臨床表型與患者表型進行細致比對.隨著二代基因測序技術的快速發展,基因檢測費用下降,在高通量測序獲得大量變異選點過程中,表型數據與基因數據的有效連接至關重要.整個流程涉及多個環節,由不同專業背景的人員和機構參與,如何確保信息的可靠性,以及在傳遞過程中避免信息丟失和發生偏倚,需建立共同語言.HPO即為很好的連接方式,可以將醫師所了解的患者表型信息、檢測機構獲得的患者基因信息、疾病和基因數據庫三者進行連接,為遺傳性疾病的診斷提供重要支持工具.
[1]Liu ZL,Liang XL,Zhang C.Neurogenetics.3rd.Beijing:People's Medical Publishing House,2011:85[.劉焯霖,梁秀齡,張成.神經遺傳病學.3版.北京:人民衛生出版社,2011:85.]
[2]Matilla?Due?as A,Sánchez I,Corral?Juan M,Dávalos A,Alvarez R,Latorre P.Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxia.Cerebellum,2010,9:148?166.
[3]Chen JW,Zhao L,Zhang F,Li L,Gu YH,Zhou JY,Zhang H,Meng M,Zhang KH,Le WD,Dong CB.Clinical characteristics,radiological features and gene mutation in 10 Chinese families with spinocerebellar ataxias.Chin Med J(Engl),2015,128:1714?1723.
[4]Serrano?Munuera C,Corral?Juan M,Stevanin G,San Nicolás H,Roig C,Corral J,Campos B,de Jorge L,Morcillo?Suárez C,Navarro A,Forlani S,Durr A,Kulisevsky J,Brice A,Sánchez I,Volpini V,Matilla?Due?as A.New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome.JAMA Neurol,2013,70:764?771.
[5]Tsoi H,Yu AC,Chen ZS,Ng NK,Chan AY,Yuen LY,Abrigo JM,Tsang SY,Tsui SK,Tong TM,Lo IF,Lam ST,Mok VC,Wong LK,Ngo JC,Lau KF,Chan TF,Chan HY.A novel missense mutation in CCDC88C activates JNK pathway and causes a dominant form of spinocerebellar ataxia.J Med Genet,2014,51:590?595.
[6]Chen KH,Wu RM,Lin HI,Tai CH,Lin CH.Mutational analysis of SYNJ1 gene(PARK20)in Parkinson's disease in a Taiwanese population.Neurobiol Aging,2015,36:2905.
[7]Kirola L,Behari M,Shishir C,Thelma BK.Identification of a novel homozygous mutation Arg459Pro in SYNJ1 gene of an Indian family with autosomal recessive juvenile parkinsonism.Parkinson Relat Disord,2016,31:124?128.
[8]Schelhaas HJ,van de Warrenburg BP.Clinical,psychological,andgenetic characteristicsofspinocerebellarataxia type 19(SCA19).Cerebellum,2005,4:51?54.
[9]Schelhaas HJ,Ippel PF,Hageman G,Sinke RJ,van der Laan EN,Beemer FA.Clinical and genetic analysis of a four?generation family with a distinct autosomal dominant cerebellar ataxia.J Neurol,2001,248:113?120.
[10]Verbeek DS,Schelhaas JH,Ippel EF,Beemer FA,Pearson PL,Sinke RJ.Identification of a novel SCA locus(SCA19)in a Dutch autosomal dominant cerebellar ataxia family on chromosome region 1p21?q21.Hum Genet,2002,111:388?393.
[11]Chung MY,Lu YC,Cheng NC,Soong BW.A novel autosomal dominant spinocerebellar ataxia(SCA22)linked to chromosome 1p21?q23.Brain,2003,126:1293?1299.
[12]Schelhaas HJ,Verbeek DS,Van de Warrenburg BP,Sinke RJ.SCA19 and SCA22:evidence for one locus with a worldwide distribution.Brain,2004,127:E6.
[13]Duarri A,Jezierska J,Fokkens M,Meijer M,Schelhaas HJ,den Dunnen WF,van Dijk F,Verschuuren?Bemelmans C,Hageman G,van de Vlies P,Küsters B,van de Warrenburg BP,Kremer B,Wijmenga C,Sinke RJ,Swertz MA,Kampinga HH,Boddeke E,Verbeek DS.Mutations in potassium channel KCND3 cause spinocerebellar ataxia type 19.Ann Neurol,2012,72:870?880.
[14]Lee YC,Durr A,Majczenko K,Huang YH,Liu YC,Lien CC,Tsai PC,Ichikawa Y,Goto J,Monin ML,Li JZ,Chung MY,Mundwiller E,Shakkottai V,Liu TT,Tesson C,Lu YC,Brice A,Tsuji S,Burmeister M,Stevanin G,Soong BW.Mutations in KCND3 cause spinocerebellar ataxia type 22.Ann Neurol,2012,72:859?869.
[15]Duarri A,Lin MC,Fokkens MR,Meijer M,Smeets CJ,Nibbeling EA,Boddeke E,Sinke RJ,Kampinga HH,Papazian DM,Verbeek DS. Spinocerebellar ataxia type 19/22 mutations alter heterocomplex Kv4.3 channel function and gating in a dominant manner.Cell Mol Life Sci,2015,72:3387?3399.
[16]Kong W,Po S,Yamagishi T,Ashen MD,Stetten G,Tomaselli GF.Isolation and characterization of the human gene encoding Ito:further diversity by alternative mRNA splicing.Am J Physiol,1998,275:H1963?1970.
[17]Isbrandt D,Leicher T,Waldschütz R,Zhu X,Luhmann U,Michel U,Sauter K,Pongs O.Gene structures and expression profiles of three human KCND(Kv4)potassium channels mediating A?type currentsⅠ(TO)andⅠ(SA).Genomics,2000,64:144?154.
[18]Dilks D,Ling HP,Cockett M,Sokol P,Numann R.Cloning and expression of the human kv4.3 potassium channel.J Neurophysiol,1999,81:1974?1977.
[19]Duarri A,Nibbeling E,Fokkens MR,Meijer M,Boddeke E,Lagrange E,Stevanin G,Brice A,Durr A,Verbeek DS.Erratum to:the L450F corrected mutation in KCND3 brings spinocerebellar ataxia and Brugada syndrome closer together.Neurogenetics,2015,16:243.
[20]De Rosa A,Pellegrino T,Pappatà S,Lieto M,Bonifati V,Palma V,Topa A,Santoro L,Bilo L,Cuocolo A,De Michele G.Non?motor symptoms and cardiac innervation in SYNJ1?related parkinsonism.Parkinsonism Relat Disord,2016,23:102?105.
[21]QuadriM,Fang M,Picillo M,OlgiatiS,Breedveld GJ,Graafland J,Wu B,Xu F,Erro R,Amboni M,Pappatà S,Quarantelli M,Annesi G,Quattrone A,Chien HF,Barbosa ER;The International Parkinsonism Genetics Network;Oostra BA,Barone P,Wang J,Bonifati V.Mutation in the SYNJ1 gene associated with autosomal recessive,early?onset parkinsonism.Hum Mutat,2013,34:1208?1215.
Clinical phenotype and genetic mutation of one case with head tremor and cerebellar atrophy
XIE Kun?ming1,GU Wei?hong2,HAO Ying2,CHEN Yuan?yuan2,ZHANG Jin2,ZHANG Xin21Grade 2014,College of Traditional Chinese Medicine,Beijing University of Chinese Medicine,Beijing 100029,China
2Movement Disorder&Neurogenetics Research Center,China?Japan Friendship Hospital,Beijing 100029,China Corresponding author:GU Wei?hong(Email:jane55.gu@vip.sina.com)
ObjectiveTo make the diagnosis for a patient presented with head tremor and cerebellar atrophy by integrating clinical features and accessory examination with genetic testing and to explore the interpretation of genetic testing results.MethodsA 30?year?old male patient's medical information,clinical pheontype,family history and accessory examinations were collected.The next?generation sequencing(NGS)of exons in 3994 causative genes of Mendelian inheritance diseases and the family tree verification were carried out.China Human Phenotype Ontology(CHPO),Phenomizer,Ensembl and Online Mendelian Inheritance in Man(OMIM)database were used to interpret the genetic test results.ResultsThe patient carried heterozygous mutation of spinocerebellar ataxia type 19(SCA19)related KCND3 gene c.1057A>G(p.Ser353Gly),but his parents did not carry this mutation.The patient also carried heterozygous mutation of parkinsonism type 20(PARK20)related SYNJ1 gene c.4436C>T(p.Thr1479Ile)which was also seen in his mother.Phenotypic similarity analysis showed the patient's phenotype was correspond with the phenotype of SCA19,and the variation locus of KCND3 gene c.1057A>G was highly conservative with homologous gene in different species.ConclusionsBy means of the integration of clinical phenotype with the result of genetic test,KCND3 gene c.1057A>G(p.Ser353Gly)carried in the patient is the pathogenic mutation.
Spinocerebellar ataxias; Phenotype; Genes; Mutation
This study was supported by Grant Awarded 2010-2012 from Ministry of Health Foundation of China.
10.3969/j.issn.1672?6731.2017.07.007
衛生部部署(管)醫院2010-2012年度臨床學科重點項目
100029北京中醫藥大學中醫學院2014級(謝坤銘);100029北京,中日友好醫院運動障礙與神經遺傳病研究中心(顧衛紅,郝瑩,陳園園,張瑾,張鑫)
顧衛紅(Email:jane55.gu@vip.sina.com)
2017?06?08)
