脂肪酸羥化酶相關性神經變性病四例臨床表型及基因突變分析
2017-11-21 11:46:26黃嘯君劉曉黎王田沈雋逸陳生弟唐維國曹立
中國現代神經疾病雜志 2017年7期
關鍵詞:基因突變
黃嘯君 劉曉黎 王田 沈雋逸 陳生弟 唐維國 曹立
脂肪酸羥化酶相關性神經變性病四例臨床表型及基因突變分析
黃嘯君 劉曉黎 王田 沈雋逸 陳生弟 唐維國 曹立
目的報道4例脂肪酸羥化酶相關性神經變性病患者,并復習相關文獻,總結該病臨床表型和基因突變特點.方法收集4例脂肪酸羥化酶相關性神經變性病患者臨床資料和家系資料,標準酚氯仿法提取患者及其父母基因組DNA并行Sanger測序.結果4例患者中3例(例2、例3和例4)具有典型脂肪酸羥化酶相關性神經變性病表現,1例(例1)表現為非典型.FA2H基因檢測顯示,4例患者均存在FA2H基因突變,其中例1為復合雜合突變c.461G>A(p.Arg154His)和c.794T>G(p.Phe265Cys);例2僅發現1種已報道的雜合突變c.703C>T(p.Arg235Cys),進一步對例2及其母進行單核苷酸多態性檢測,亦未發現缺失突變;例3為雜合突變c.688G>A(p.Glu230Lys)和插入突變c.172_173insGGGCCAGGAC(p.Ile58ArgfsX47);例4為復合雜合突變c.688G>A(p.Glu230Lys)、c.968C>A(p.Pro323Gln)和c.976G>A(p.Gly326Asp),其父為c.688G>A(p.Glu230Lys)突變攜帶者,其母為c.968C>A(p.Pro323Gln)和c.976G>A(p.Gly326Asp)突變攜帶者.根據美國醫學遺傳學和基因組學會標準,例1的FA2H基因雜合突變c.461G>A(p.Arg154His)為"可能致病"、c.794T>G(p.Phe265Cys)為"可能致病";例2的FA2H基因雜合突變c.703C>T(p.Arg235Cys)為"可能致病";例3的FA2H基因雜合突變c.688G>A(p.Glu230Lys)為"致病"、插入突變c.172_173insGGGCCAGGAC(p.Ile58ArgfsX47)為"致病";例4的FA2H基因雜合突變c.688G>A(p.Glu230Lys)為"致病"、c.968C>A(p.Pro323Gln)為"致病"、c.976G>A(p.Gly326Asp)為"可能致病".結論脂肪酸羥化酶相關性神經變性病具有高度臨床和遺傳異質性,痙攣性截癱是最主要的臨床表現,對于復雜型常染色體隱性遺傳性遺傳性痙攣性截癱,尤其合并構音障礙、智力減退、腦白質病變和小腦萎縮等臨床特征的患者,應考慮FA2H基因突變導致的脂肪酸羥化酶相關性神經變性病.
神經變性疾病; 脂肪酸類; 混合功能氧化酶類; 表型; 基因; 突變
FA2H基因突變與腦白質營養不良、遺傳性痙攣性截癱35型(SPG35型)和腦組織鐵沉積性神經變性病(NBIA)均相關[1?3].該基因突變導致的腦組織鐵沉積性神經變性病亞型占所有腦組織鐵沉積性神經變性病的比例較低(<1%),臨床表現具有一定特異性,典型表現為痙攣性截癱、小腦共濟失調、認知功能障礙、癲發作等.FA2H基因突變導致的臨床表型存在相互重疊,故將上述3種臨床表型統稱為脂肪酸羥化酶相關性神經變性病[FAHN,在線人類孟德爾遺傳數據庫(OMIM)編號:611026][1].本研究報道4例脂肪酸羥化酶相關性神經變性病患者,并復習相關文獻,總結該病臨床表型和基因突變特點.
對象與方法
一、研究對象
研究對象均來自2012年1月-2017年1月上海交通大學醫學院附屬瑞金醫院神經科擬診脂肪酸羥化酶相關性神經變性病患者,診斷依據臨床表型,其中表現為痙攣性截癱表型的患者,參照Harding診斷標準[4];共4例,男性3例,女性1例;年齡5~50歲,平均28歲.以及4例患者父母共7例(例2父親已死亡),男性3例,女性4例;年齡30~78歲,平均56.14歲;均無相似臨床癥狀,否認近親婚配.本研究經上海交通大學醫學院附屬瑞金醫院道德倫理委員會審核批準,所有患者及其家屬均知情同意并簽署知情同意書.
二、研究方法
1.一般資料收集 采集4例患者的家系資料、臨床病史,以及神經系統查體、影像學檢查等.
2.標本收集與基因組DNA提取 采集4例患者及其父母外周靜脈血各5 ml,予乙二胺四乙酸(EDTA)抗凝管(美國BD公司)抗凝.采用標準酚氯仿法提取患者及其父母基因組DNA,紫外分光光度計(美國Bio?Rad公司)于260和280 nm波長處定量測定基因組DNA.
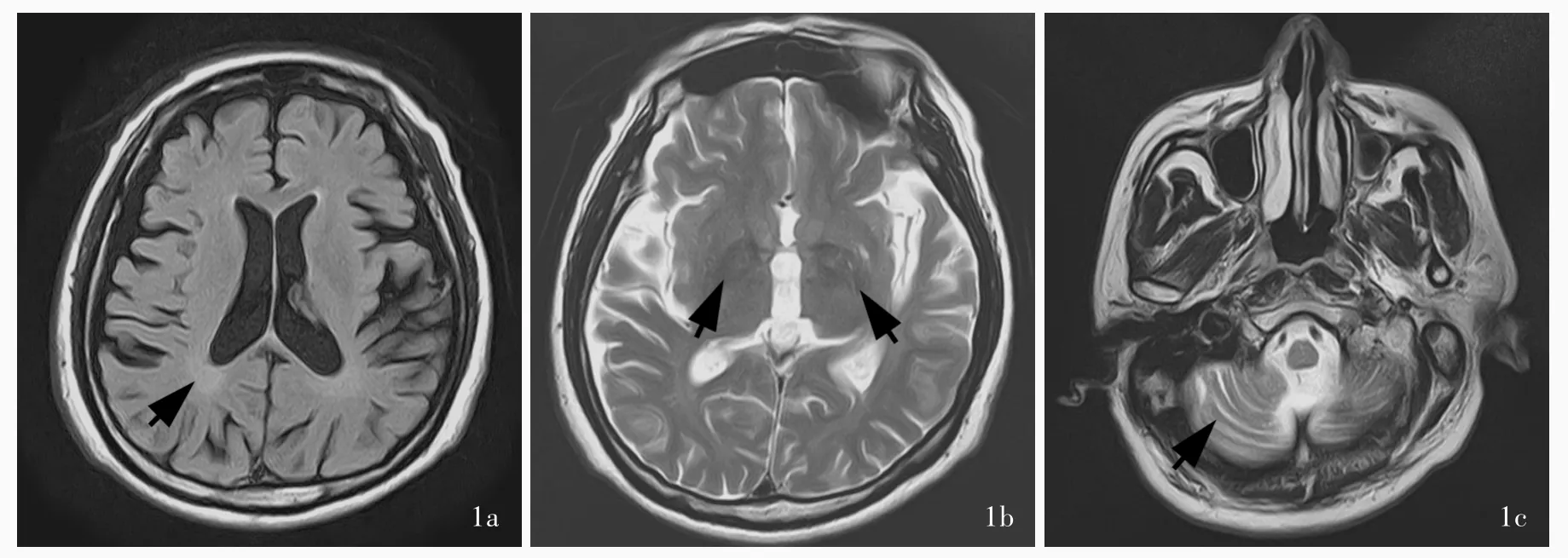
圖1 例1頭部MRI檢查所見 1a 橫斷面FLAIR成像顯示,雙側側腦室旁稍高信號影(箭頭所示),提示腦白質病變1b 橫斷面T2WI顯示,雙側基底節區鐵離子沉積(箭頭所示)1c 橫斷面 T2WI顯示,小腦腦溝輕度增寬(箭頭所示)Figure 1 Head MRI findings in Case 1 Axial FLAIR showed bilateral paraceles high?intensity signal,suggesting lesion in white matter of the brain(arrow indicates,Panel 1a).Axial T2WI reveald ferrum deposit in bilateral basal ganglia regions(arrows indicate,Panel 1b).Axia T2WI indicated cerebellar sulus slightly broaden(arrow indicates,Panel 1c).
3.Sanger測序 (1)聚合酶鏈反應(PCR):根據FA2H基因外顯子進行PCR擴增,參照FA2H基因第1~7外顯子及其側翼序列設計引物,由英濰捷基(上海)貿易有限公司合成,正向引物序列為FA2H?1F:5'?GGTATGCAAATGAGCAGGTG?3'、FA2H?2F:5'?CTTGACCTCCCAAAGTGCTG ?3'、FA2H ?3F:5'?CCTGCCGATTTAGCACCTC ?3'、FA2H ?4F:5'?GGCTGAGGCAGAAGAGTCAC ?3'、FA2H ?5F:5'?AAGGACCCCAGCAGTATGG ?3'、FA2H ?6F:5'?ACCAAAGGTGCCAAGCCTAC ?3'、FA2H ?7F:5'?GCTGGGGACAATAAAAGACAC?3',反向引物序列為 FA2H?1R:5'?GAGAGAGGAAGGAGGGGTTG?3'、FA2H ?2R:5'?TGGACACCTGCTTCTCCTTC ?3'、FA2H?3R:5'?ATGTTCCTCCCTCCCTGAC?3'、FA2H?4R:5'?TTTCTGCTGATGTGTTGTTGG?3'、FA2H?5R:5'?AAAGGTGATGCGTAAACAAGG?3'、FA2H?6R:5'?GTCTGCACCAGGGAAAGAG ?3'、FA2H ?7R:5'?TGGATGTGACCCTCCTACC?3'.PCR反應體系共25 μl,依次加入 dNTPs 2.50 mmol、2XGC 緩沖液Ⅰ12.50 μl、各引物序列 5 pmol、模板 DNA 100 ng以及r?Taq 1 U,再加水補充至25 μl;反應條件為 95 ℃預變性5 min,95℃ 90 s、62 ℃ 1 min、72℃ 2 min,共循環35次,72℃延伸10 min.(2)Sanger測序:PCR擴增產物采用DNAStar軟件包中SeqMan軟件(美國DNAStar公司)進行比對.例2及其母(其父已死亡,未能采集標本)基因組DNA采用Illumina Human Ommi Zhonghua?8 Bead Chips全基因組分型芯片(美國Illumina公司)進行單核苷酸多態性(SNP)檢測.對檢出的FA2H基因突變,根據美國醫學遺傳學和基因組學會(ACMG)標準[5]進行致病性判定.
結 果
4例患者臨床表現符合脂肪酸羥化酶相關性神經變性病,經基因檢測確定存在FA2H基因突變.
一、臨床特征
例1男性,50歲,因進行性行走困難16年,于2014年6月5日至我院神經科門診就診.患者16年前(34歲時)無明顯誘因開始出現行走不穩,逐漸進展為雙下肢乏力,伴僵硬感,進行性加重,同時伴言語欠流利,但無明顯飲水嗆咳,上肢活動自如,精細動作尚可準確完成,無肢體抽搐、記憶力減退;3年前開始出現排尿控制障礙.患者生長發育里程碑與同齡正常人無明顯差異,智力發育可.父母無相似臨床癥狀或其他神經系統疾病,否認近親婚配.門診神經系統檢查:神志清楚,語言欠流利;雙側瞳孔等大、等圓,直徑約3 mm,對光反射靈敏;雙側鼻唇溝對稱,伸舌居中;雙下肢髂腰肌肌力4+級、股四頭肌5級、脛前肌5級、腓腸肌5級,雙側外展肌肌力4+級、內收肌5級,雙下肢肌張力略增高;剪刀步態;雙側快復輪替動作笨拙,指鼻試驗和跟?膝?脛試驗尚可,Romberg征陽性;雙上肢腱反射正常、雙下肢膝反射和踝反射增強、雙側踝陣攣減弱,雙側病理征陽性.影像學檢查:頭部MRI顯示輕度小腦萎縮,胼胝體無明顯萎縮,雙側側腦室角白質變性,雙側基底節區鐵離子沉積(圖1).
例2女性,22歲,因進行性行走困難19年,發作性意識喪失伴肢體抽搐3年,于2012年9月5日至我院神經科門診就診.患者19年前(3歲時)開始出現行走困難,伴下肢僵硬感,進行性加重,隨后出現智力減退;小學3年級(10歲)時輟學,隨后逐漸出現雙手動作笨拙、言語模糊,但無吞咽困難和飲水嗆咳;19歲時出現癲發作,一直服用丙戊酸鈉至今;目前已喪失獨立行走能力,大小便控制障礙.患者出生至學齡前生長發育無明顯異常.父母無相似臨床癥狀或其他神經系統疾病,否認近親婚配.門診神經系統檢查:神志清楚,語言欠流利;雙側瞳孔等大、等圓,直徑約3 mm,對光反射靈敏;雙側鼻唇溝對稱,伸舌居中;雙上肢肌力5級、雙下肢3-級,四肢肌張力增高;雙側快復輪替動作笨拙,步態檢查不能完成;雙上肢腱反射增強、雙下肢膝反射強陽性和踝反射增強、踝陣攣減弱,雙側病理征陽性.頭部MRI顯示腦白質病變.
例3男性,5歲,主因進行性行走困難2年,于2016年4月13日至我院神經科門診就診.患兒2年前(3歲時)開始出現行走困難,行走時雙腿內收,進行性加重,伴語速減慢,雙上肢無明顯活動異常.患兒生長發育與同齡正常兒童無明顯差異,智力發育可.父母無相似癥狀或其他神經系統疾病,否認近親婚配.門診神經系統檢查:神志清楚,語速緩慢;雙側瞳孔等大、等圓,直徑約3 mm,對光反射靈敏;雙側鼻唇溝對稱,伸舌居中;雙上肢肌力5級、雙下肢4級,雙下肢內收肌力5級、外展肌力3級,四肢肌張力增高;呈剪刀步態;雙側指鼻試驗尚可,Romberg征陰性;雙上肢腱反射正常,雙下肢膝反射和踝反射增強、雙側踝陣攣減弱,雙側病理征陽性.頭部MRI顯示雙側側腦室后角旁腦白質病變,小腦延髓池輕度增寬.
例4男性,35歲,主因進行性行走困難31年,發聲困難、飲水嗆咳19年,發作性意識喪失伴肢體抽搐10年,于2012年6月15日至我院神經科門診就診.患者31年前(4歲時)出現雙側足背屈困難,行走時足尖著地,長時間行走后雙側膝關節疼痛,進行性加重;5歲時出現行走困難,伴左足不自主內翻,進行性加重;6歲時出現握筆姿勢異常,表現為內旋,書寫尚工整;8歲(小學1年級)時學習成績尚可,但發聲低、書寫慢;9歲(小學2年級下半學期,留級1年)學習成績落后,行走姿勢異常進一步加重,雙側膝關節不能彎曲;12歲(小學5年級時)右足不自主內翻,不能獨立行走,行矯形術;小學畢業后輟學;16歲時不能行走并出現發聲困難、飲水嗆咳;20歲時雙手呈握拳姿勢,語速明顯減慢,需使用輪椅;25歲時曾出現一次午睡后一側肢體和口角不自主抽搐,眼球向同側凝視伴意識喪失,持續5 min后好轉,此后反復出現類似發作,持續30 min以上,服用丙戊酸鈉后癥狀控制欠佳,進行性加重,逐漸出現言語困難,只能講單字,保持坐位困難,大小便失禁;28歲時完全不能言語,僅能點頭示意,此后逐漸出現頭頸部無力,頭向后仰或偏向左側,僅能眨眼示意;30歲時反應遲鈍,呼之反應差;32歲后抽搐發作逐漸減少,自行停用丙戊酸鈉,家人訴其肢體僵硬感較前略有好轉.患者父母無相似癥狀或其他神經系統疾病,否認近親婚配;其妹5歲時開始出現相似臨床癥狀.門診體格檢查:神志清楚,言語不能,反應遲鈍,計算力和定向力差;雙側瞳孔等大、等圓,直徑約為4 mm,直接和間接對光反應靈敏,余腦神經檢查不配合;四肢肌肉極度萎縮,肌力0級,雙上肢關節屈曲攣縮,雙下肢肌張力降低;行走不能;四肢腱反射未引出,雙側病理征陰性.頭部MRI顯示,大腦皮質、胼胝體和小腦萎縮,伴腦白質營養不良和基底節區T2WI低信號.
二、基因檢測
FA2H基因檢測顯示,4例患者均存在FA2H基因突變,例1為復合雜合突變c.461G>A(p.Arg154His)和c.794T>G(p.Phe265Cys,圖2),此2種突變在家系中出現共分離現象;例2僅發現1種已報道的雜合突變c.703C>T(p.Arg235Cys,圖3),其母未攜帶該突變,進一步對例2及其母進行單核苷酸多態性檢測,亦未發現FA2H基因缺失突變;例3為雜合突變c.688G>A(p.Glu230Lys)和插入突變c.172_173insGGGCCAGGAC(p.Ile58ArgfsX47,圖4),此2種突變在家系中出現共分離現象;例4為復合雜合突變c.688G>A(p.Glu230Lys)、c.968C>A(p.Pro323Gln)和c.976G>A(p.Gly326Asp,圖5),其父為c.688G>A(p.Glu230Lys)突變攜帶者,其母為c.968C>A(p.Pro323Gln)和 c.976G>A(p.Gly326Asp)突變攜帶者.根據美國醫學遺傳學和基因組學會標準[5]進行致病性突變判定,例1的FA2H基因雜合突變c.461G>A(p.Arg154His)為"可能致病"、c.794T>G(p.Phe265Cys)為"可能致病";例2的FA2H基因雜合突變c.703C>T(p.Arg235Cys)為"可能致病";例3的FA2H基因雜合突變c.688G>A(p.Glu230Lys)為"致病"、插入突變c.172_173insGGGCCAGGAC(p.Ile58ArgfsX47)為"致病";例4的FA2H基因雜合突變c.688G>A(p.Glu230Lys)為"致病"、c.968C>A(p.Pro323Gln)為"致病"、c.976G>A(p.Gly326Asp)為"可能致病".

圖2 例1 Sanger測序顯示,FA2H基因存在復合雜合突變c.461G>A(p.Arg154His,箭頭所示,左圖)和c.794T>G(p.Phe265Cys,箭頭所示,右圖)Figure 2 Sanger sequencing in Case 1 showed FA2H gene compound heterozygous mutation c.461G>A (p.Arg154His,arrow indicates,left panel)and c.794T>G(p.Phe265Cys,arrow indicates,right panel).
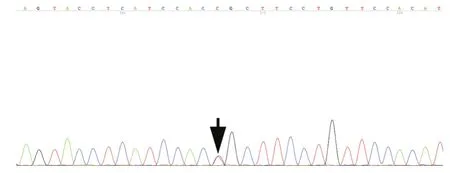
圖3 例2 Sanger測序顯示,FA2H基因存在雜合突變c.703C>T(p.Arg235Cys,箭頭所示)Figure 3 Sanger sequencing in Case 2 showed FA2H genecompound heterozygousmutation c.703C>T (p.Arg235Cys,arrow indicates).
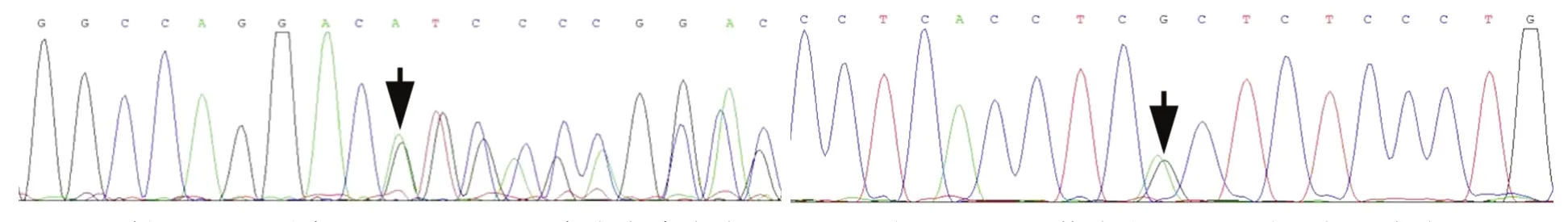
圖4 例3 Sanger測序顯示,FA2H基因存在雜合突變c.688G>A(p.Glu230Lys,箭頭所示,左圖)和插入突變c.172_173insGGGCCAGGAC(p.Ile58ArgfsX47,箭頭所示,右圖)Figure 4 Sanger sequencing in Case 3 showed FA2H gene compound heterozygous mutation c.688G>A(p.Glu230Lys,arrow indicaties,left panel)and insertion mutation c.172_173insGGGCCAGGAC(p.Ile58ArgfsX47,arrow indicates,right panel).

圖5 例4 Sanger測序顯示,FA2H基因存在復合雜合突變c.688G>A(p.Glu230Lys,箭頭所示,左圖)、c.968C>A(p.Pro323Gln,箭頭所示,右圖)和c.976G>A(p.Gly326Asp,箭頭所示,右圖)Figure 5 Sanger sequencing in Case 4 showed FA2H gene compound heterozygous mutation c.688G>A(p.Glu230Lys,arrow indicates,left panel),c.968C>A(p.Pro323Gln,arrow indicates,right panel)and c.976G>A(p.Gly326Asp,arrow indicates,right panel).
討 論
本研究報道4例FA2H基因突變導致的脂肪酸羥化酶相關性神經變性病患者.FA2H基因編碼FA2H蛋白,包含372個氨基酸,主要作用是催化脂肪酸N?酰基鏈羥化.FA2H蛋白是神經酰胺前體,是髓鞘形成關鍵成分[6].由于磷脂半乳糖脂肪酸在髓鞘脂肪成分中占1/3[7],因此,FA2H蛋白對維持髓鞘穩定性具有重要作用.FA2H基因突變導致FA2H蛋白失活引起羥化作用缺失而影響正常髓鞘形成.異常的髓鞘形成可能促使神經元功能障礙和凋亡.FA2H基因突變曾被認為與腦白質病[2]、SPG35型[3]以及腦組織鐵沉積神經變性病相關[1].由于此3種亞型臨床表現存在重疊,故統稱為脂肪酸羥化酶相關性神經變性病[8],表現為痙攣性截癱、肌張力障礙、癲發作、視神經萎縮、智力減退、小腦萎縮、腦白質病變和腦組織鐵離子沉積等.目前全球僅有 56 例報道[2?3,8?19].
本研究4例患者均于幼年時期出現痙攣性截癱,而總結已報道的脂肪酸羥化酶相關性神經變性病病例,所有患者均存在痙攣性步態,且為首發癥狀,因此痙攣性截癱是脂肪酸羥化酶相關性神經變性病最常見和最突出的亞型,既往將FA2H基因突變導致的痙攣性截癱定義為SPG35型,系復雜型遺傳性痙攣性截癱,常合并肌張力障礙、共濟失調、構音障礙、智力減退和癲發作等臨床癥狀.本研究有3例患者(例2、例3和例4)符合復雜型遺傳性痙攣性截癱臨床表現,而例1臨床表型為緩慢進展的晚發型單純型遺傳性痙攣性截癱,無智力減退和癲發作,頭部MRI顯示輕度腦白質病變、小腦萎縮和腦組織鐵離子沉積,無明顯胼胝體萎縮,從而拓展SPG35型和脂肪酸羥化酶相關性神經變性病表型譜.值得注意的是,隨著分子檢測技術的普及,已有數項研究報道非典型SPG35型或脂肪酸羥化酶相關性神經變性病[11,16],其共同點是發病年齡較晚,癥狀較輕,進展緩慢,影像學無腦白質病變或腦組織鐵離子沉積,提示在常染色體隱性遺傳性遺傳性痙攣性截癱中,FA2H基因突變導致的SPG35型或脂肪酸羥化酶相關性神經變性病并不少見.近期在漢族復雜型常染色體隱性遺傳性遺傳性痙攣性截癱研究中發現,SPG35型發病率為2.32%,僅次于SPG11型(11.62%)[12].另一項針對合并胼胝體萎縮、智力減退或腦白質病變的復雜型常染色體隱性遺傳性遺傳性痙攣性截癱研究顯示,SPG35型發病率(4.91%)亦低于SPG11型(26.22%),但高于SPG48型(3.27%)[11].提示對于復雜型常染色體隱性遺傳性遺傳性痙攣性截癱患者,應考慮SPG35型或脂肪酸羥化酶相關性神經變性病的可能,而不僅局限于合并癲發作、認知功能障礙、腦白質病變、腦組織鐵離子沉積的遺傳性痙攣性截癱.
此外,總結文獻報道的56例脂肪酸羥化酶相關性神經變性病患者發現,脂肪酸羥化酶相關性神經變性病中癲發作發生率僅為29.09%,提示癲發作可能是脂肪酸羥化酶相關性神經變性病的特征性表現但不具有普遍性,而構音障礙、智力減退發生率較高.此外,脂肪酸羥化酶相關性神經變性病影像學表現為腦白質病變及小腦和腦干萎縮,與臨床表現上的構音障礙和智力減退相符,而腦組織鐵離子沉積和胼胝體萎縮相對少見.因此,對于存在構音障礙、智力減退同時伴腦白質病變、小腦萎縮的遺傳性痙攣性截癱患者,應考慮脂肪酸羥化酶相關性神經變性病的可能.
本研究有2例患者(例3和例4)臨床表現為典型脂肪酸羥化酶相關性神經變性病表現,例3目前年齡尚小,是否會出現癲發作、認知功能障礙尚待進一步隨訪觀察.該2例患者FA2H基因突變根據美國醫學遺傳學和基因組學會標準[5]均判定為"致病",且例3為插入突變,該突變可以導致終止密碼子提前出現,從而導致蛋白截短或蛋白功能缺失,由此推測例3臨床表現可能與典型脂肪酸羥化酶相關性神經變性病相符.例4存在3種FA2H基因雜合突變,共分離研究顯示,c.688G>A(p.Glu230Lys)位于一條染色體,c.968C>A(p.Pro323Gln)和c.976G>A(p.Gly326Asp)則位于另一條染色體,根據脂肪酸羥化酶相關性神經變性病呈常染色體隱性遺傳的特性,一般認為c.968C>A(p.Pro323Gln)和c.976G>A(p.Gly326Asp)中的一個為致病性突變位點,根據美國醫學遺傳學和基因組學會標準[5]判定c.968C>A(p.Pro323Gln)為"致病",而c.976G>A(p.Gly326Asp)為"可能致病",因此傾向c.968C>A(p.Pro323Gln)為致病性突變,仍不能排除2種突變均造成酶功能改變而致病的可能,尚待進一步功能學試驗證實.例1臨床表型為非典型脂肪酸羥化酶相關性神經變性病,存在FA2H基因復合雜合突變c.461G>A(p.Arg154His)和 c.794T>G(p.Phe265Cys),均為未報道的錯義突變,對蛋白功能影響尚不明確,但2個突變位點高度保守,而與c.461G>A(p.Arg154His)位點相鄰、導致相同氨基酸位點改變的純合突變c.460C>T(p.Arg154Cys)曾被報道導致相同疾病的發生[1],故根據美國醫學遺傳學和基因組學會標準[5]判斷為"可能致病",上述2種突變對酶活性的影響尚待進一步研究.由于例1患者表現為非典型脂肪酸羥化酶相關性神經變性病,我們推測復合雜合錯義突變導致酶活性部分下降可能是潛在原因;而部分非典型脂肪酸羥化酶相關性神經變性病患者腦白質病變程度較輕或不存在腦白質病變[16],可能與酶活性部分保留可維持部分髓鞘磷脂形成有關[20],尚待進一步功能學試驗驗證.例2患者表型符合脂肪酸羥化酶相關性神經變性病的特征性表現,主要表現為幼年期發病的痙攣性截癱、智力減退、癲發作等,但FA2H基因檢測僅發現1種已報道的雜合突變c.703C>T(p.Arg235Cys),對其母進行FA2H基因檢測未發現該突變,推測該突變源自其父,進一步的單核苷酸多態性檢測未能發現患者及其母存在缺失突變,因此是否存在其他遺傳情況以及單一突變如何導致疾病尚不明確,由于脂肪酸羥化酶相關性神經變性病為常染色體隱性遺傳,故單一突變可能會引起酶羥化功能降低而導致臨床癥狀的出現,尚待進一步對該位點進行功能學試驗證實.
綜上所述,本研究報道4例脂肪酸羥化酶相關性神經變性病患者.脂肪酸羥化酶相關性神經變性病表型多樣,但痙攣性截癱是其最主要的臨床表現,對于復雜型常染色體隱性遺傳性遺傳性痙攣性截癱,尤其合并構音障礙、智力減退、腦白質病變和小腦萎縮等臨床特征的患者,應考慮FA2H基因導致的脂肪酸羥化酶相關性神經變性病.
[1]Kruer MC,Paisán ?Ruiz C,Boddaert N,Yoon MY,Hama H,Gregory A,Malandrini A,Woltjer RL,Munnich A,Gobin S,Polster BJ,Palmeri S,Edvardson S,Hardy J,Houlden H,Hayflick SJ.Defective FA2H leads to a novelform of neurodegeneration with brain iron accumulation(NBIA).Ann Neurol,2010,68:611?618.
[2]Edvardson S,Hama H,Shaag A,Gomori JM,Berger I,Soffer D,Korman SH,Taustein I,Saada A,Elpeleg O.Mutations in the fatty acid 2?hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia.Am J Hum Genet,2008,83:643?648.
[3]Dick KJ,Eckhardt M,Paisán?Ruiz C,Alshehhi AA,Proukakis C,Sibtain NA,Maier H,Sharifi R,Patton MA,Bashir W,Koul R,Raeburn S,Gieselmann V,Houlden H,Crosby AH.Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia(SPG35).Hum Mutat,2010,31:E1251?1260.
[4]Harding AE.Classification ofthe hereditary ataxias and paraplegias.Lancet,1983,1:1151?1155.
[5]Richards S,Aziz N,Bale S,Bick D,Das S,Gastier?Foster J,Grody WW,Hegde M,Lyon E,Spector E,Voelkerding K,Rehm HL;ACMG Laboratory Quality Assurance Committee.Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.Genet Med,2015,17:405?424.
[6]Eckhardt M,Yaghootfam A,Fewou SN,Z?ller I,Gieselmann V.A mammalian fatty acid hydroxylase responsible for the formation of alpha?hydroxylated galactosylceramide in myelin.Biochem J,2005,388:245?254.
[7]Morell P,Greenfield S,Norton WT,Wisniewski H.Isolation and characterization of myelin protein from adult quaking mice and its similarity to myelin protein of young normal mice.Adv Exp Med Biol,1972,32:251?261.
[8]Pierson TM,Simeonov DR,Sincan M,Adams DA,Markello T,Golas G,Fuentes?Fajardo K,Hansen NF,Cherukuri PF,Cruz P,Mullikin JC,Blackstone C,Tifft C,Boerkoel CF,Gahl WA;NISC Comparative Sequencing Program.Exome sequencing and SNP analysis detect novel compound heterozygosity in fatty acid hydroxylase?associated neurodegeneration.Eur J Hum Genet,2012,20:476?479.
[9]Cao L,Huang XJ,Chen CJ,Chen SD.A rare family with hereditary spastic paraplegia type 35 due to novelFA2H mutations:a case report with literature review.J Neurol Sci,2013,329:1?5.
[10]Garone C,Pippucci T,Cordelli DM,Zuntini R,Castegnaro G,Marconi C,GrazianoC,Marchiani V,Verrotti A,SeriM,Franzoni E.FA2H?related disorders:a novel c.270+3A>T splice?site mutation leads to acomplexneurodegenerative phenotype.Dev Med Child Neurol,2011,53:958?961.
[11]Pensato V,Castellotti B,Gellera C,Pareyson D,Ciano C,Nanetti L,Salsano E,Piscosquito G,Sarto E,Eoli M,Moroni I,Soliveri P,Lamperti E,Chiapparini L,Di Bella D,Taroni F,Mariotti C. Overlapping phenotypes in complex spastic paraplegias SPG11,SPG15,SPG35 and SPG48.Brain,2014,137:1907?1920.
[12]Liao X,Luo Y,Zhan Z,Du J,Hu Z,Wang J,Guo J,Hu Z,Yan X,Pan Q,Xia K,Tang B,Shen L.SPG35 contributes to the secondcommonsubtypeofAR?HSP inChina:frequency analysis and functional characterization of FA2H gene mutations.Clin Genet,2015,87:85?89.
[13]Rupps R,Hukin J,Balicki M,Mercimek?Mahmutoglu S,Rolfs A,Dias C.Novelmutations in FA2H ?associated neurodegeneration: an underrecognized condition? J Child Neurol,2013,28:1500?1504.
[14]DonkervoortS,DastgirJ,Hu Y,Zein WM,Marks H,Blackstone C,B?nnemann CG.Phenotypic variability of a likely FA2H founder mutation in a family with complicated hereditary spastic paraplegia.Clin Genet,2014,85:393?395.
[15]Zaki MS,Selim L,Mansour L,Mahmoud IG,Fenstermaker AG,Gabriel SB,Gleeson JG.Mutations in FA2H in three Arab familieswith aclinicalspectrum ofneurodegeneration and hereditary spastic paraparesis.Clin Genet,2015,88:95?97.
[16]Tonelli A,D'Angelo MG,Arrigoni F,Brighina E,Arnoldi A,Citterio A, Bresolin N,Bassi MT. Atypical adult onset complicated spastic paraparesis with thin corpus callosum in two patients carrying a novel FA2H mutation.Eur J Neurol,2012,19:E127?129.
[17]Aguirre?Rodríguez FJ,Lucenilla MI,Alvarez?Cubero MJ,Mata C,Entrala?BernalC,Fernandez?RosadoF.NovelFA2H mutation in a girl with familial spastic paraplegia.J Neurol Sci,2015,357:332?334.
[18]Magariello A,Russo C,Citrigno L,Züchner S,Patitucci A,Mazzei R,Conforti FL,Ferlazzo E,Aguglia U,Muglia M.Exome sequencing reveals two FA2H mutations in a family with a complicated form of hereditary spastic paraplegia and psychiatric impairments.J Neurol Sci,2017,372:347?349.
[19]Soehn AS,Rattay TW,Beck?W?dl S,Sch?ferhoff K,Monk D,D?bler?Neumann M,H?rtnagel K,Schlüter A,Ruiz M,Pujol A,Züchner S,Riess O,Schüle R,Bauer P,Sch?ls L.Uniparental disomy ofchromosome 16 unmasksrecessive mutationsof FA2H/SPG35 in 4 families.Neurology,2016,87:186?191.
[20]Alderson NL,Rembiesa BM,Walla MD,Bielawska A,Bielawski J,Hama H.The human FA2H gene encodes a fatty acid 2?hydroxylase.J Biol Chem,2004,279:48562?48568.
Clinicalphenotype and genetic mutation offatty acid hydroxylase?associated neurodegeneration:analysis of four cases
HUANG Xiao?jun1,LIU Xiao?li2,WANG Tian2,SHEN Jun?yi2,CHEN Sheng?di2,TANG Wei?guo3,CAO Li21Department of Neurology,North Department of Ruijin Hospital,School of Medicine,Shanghai Jiaotong University,Shanghai 201801,China
2Department of Neurology and Institute of Neurology,Ruijin Hospital,School of Medicine,Shanghai Jiaotong University,Shanghai 200025,China
3Department of Neurology,Zhoushan Hospital,Zhoushan 316021,Zhejiang,China
CAO Li(Email:caoli2000@yeah.net)
ObjectiveTo report 4 cases of fatty acid hydroxylase?associated neurodegeneration(FAHN)and tosummarizethe clinicaland geneticcharacteristicsofFAHN byliteraturesreview.MethodsFour cases of FAHN patients'clinical and family data were collected in detail.The gDNA of patients and their parents were extracted from peripheral blood.FA2H gene was conducted and followed by Sanger sequencing.ResultsAmong the 4 cases,3 cases(Case 2,Case 3,Case 4)presented typical manifestations of FAHN while the other(Case 1)was atypical.Genetic sequencing showed FA2H gene mutation in all affected patients.Compound heterozygous mutation c.461G>A(p.Arg154His)and c.794T>G(p.Phe265Cys)were seen in Case 1.In Case 2,only one documented heterozygous mutation c.703C>T(p.Arg235Cys)was found,and dificit mutation was not found in single nucleotide polymorphism(SNP)chip test of the patient and her mother. Compound heterozygous mutation c.688G>A (p.Glu230Lys)and insertion mutation c.172_173insGGGCCAGGAC(p.Ile58ArgfsX47)were presented in Case 3.In Case 4,compound heterozygous mutation c.688G>A(p.Glu230Lys),c.968C>A(p.Pro323Gln)and c.976G>A(p.Gly326Asp)were seen,while his father was the carrier of c.688G>A(p.Glu230Lys)mutation and his mother was the carrier of c.968C>A(p.Pro323Gln)and c.976G>A(p.Gly326Asp)mutation.According to the standard of American College of Medical Genetics and Genomics(ACMG),c.461G>A(p.Arg154His)and c.794T>G(p.Phe265Cys)in Case 1,and c.703C>T(p.Arg235Cys)in Case 2 were considered as"likely pathogenic",while FA2H gene compound heterozygous mutation c.688G>A(p.Glu230Lys),insertion mutation c.172_173insGGGCCAGGAC(p.Ile58ArgfsX47)in Case 3 was as"pathogenic",and in Case 4,the FA2H gene mutation c.688G>A(p.Glu230Lys)and c.968C>A(p.Pro323Gln)were"pathogenic"and c.976G>A(p.Gly326Asp)was"likely pathogenic".ConclusionsFAHN has highly clinical and genetic heterogenieity in which spastic paraplegia is the main clinical presentation.In typing diagnosis for patietns with autosomal recessive herditary spastic paraplegia(HSP),especially combined with dyslalia,dysnoesia,and clinical features of white matter lesion and cerebellar atrophy,FA2H gene mutation?induced FAHN should be considered.
Neurodegenerative diseases; Fatty acids; Mixed function oxygenases; Phenotype;Genes; Mutation
10.3969/j.issn.1672?6731.2017.07.010
國家自然科學基金資助項目(項目編號:81571086);國家自然科學基金青年科學基金資助項目(項目編號:81600978);上海交通大學醫學院高峰高原計劃(項目編號:20161401);上海交通大學"醫工交叉研究基金"資助項目(項目編號:YG2016MS64);浙江省科學技術廳2014年度公益性技術應用研究計劃項目(項目編號:2014C33132)
201801上海交通大學醫學院附屬瑞金醫院北院神經內科(黃嘯君);200025上海交通大學醫學院附屬瑞金醫院神經科 上海交通大學醫學院神經病學研究所(劉曉黎,王田,沈雋逸,陳生弟,曹立);316021浙江省舟山醫院神經內科(唐維國)
曹立(Email:caoli2000@yeah.net)
This study was supported by the National Natural Science Foundation of China(No.81571086),the National Natural Science Foundation of China for Young Scientists(No.81600978),Shanghai Jiaotong University School of Medicine Peak and Plateau Program (No.20161401),Crossing Program between Medicine and Industry supported by Shanghai Jiaotong University(No.YG2016MS64),and Non?Profit Technical Application Plan Project of Science Technology Department of Zhejiang Province,China in the Year 2014(No.2014C33132).
2017?06?24)
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
