次氯酸鈉氧化—磷酸銨鎂沉淀法處理草甘膦廢水并回收磷
2018-01-08 09:10:47李永峰朱利軍賀子良劉銳平
化工環保 2017年6期
關鍵詞:實驗
李永峰,王 歡,,朱利軍 ,賀子良,劉銳平
(1. 東北林業大學 林學院,黑龍江 哈爾濱 150040;2. 中國科學院 生態環境研究中心飲用水科學與技術重點實驗室,北京 100085)
草甘膦作為一種廣譜、低毒、高效的內吸傳導性除草劑被廣泛應用。我國是草甘膦生產和出口大國,產能已接近1 Mt/a[1]。草甘膦生產普遍采用甘氨酸法、亞氨基二乙酸法工藝[2],會產生大量磷濃度高、難降解的廢水。另一方面,磷礦作為不可再生資源日益枯竭,而磷需求量逐年穩步提高。因此,含磷廢水處理由傳統的“除磷”模式轉向“磷回收”模式勢在必行。
“磷回收”具有較好的技術經濟可行性[3]。如果農藥廠廢水中的草甘膦直接進入生化系統則可能與微生物體內的DNA、RNA和蛋白質反應,顯著抑制污泥活性[4]。篩選馴化草甘膦降解菌株有望提高其對毒性的耐受性,但要實現規模化應用仍有許多限制因素[5]。吸附[6]、沉淀[7]、Fenton氧化[8]等物理化學方法存在吸附劑再生困難、污泥量過高等不利因素。草甘膦分子較不穩定,容易被氧化生成磷酸鹽[9],而次氯酸鈉具有較強的氧化性[10],無污泥產生,且成本較低,具有應用前景。將草甘膦轉化為無機磷后再進行回收,可避免對生化系統的影響,且具有很好的經濟價值。研究發現磷酸銨鎂沉淀法可有效回收廢水中的磷酸鹽[11-13]。磷酸銨鎂中磷含量高達51.8%(以P2O5質量分數計),是優質的緩釋肥。
本研究采用次氯酸鈉氧化草甘膦得到磷酸鹽,再采用磷酸銨鎂沉淀法回收磷,考察了次氯酸鈉加入量、溶液pH、氧化時間等工藝條件對草甘膦氧化效果的影響,探討了草甘膦廢水處理與磷回收的可行性。
1 實驗部分
1.1 材料、試劑和儀器
草甘膦溶液(純度95%,美國阿達瑪斯公司提供),pH = 4.93,質量濃度16.38 g/L。實驗用水為超純水。二氯甲烷為色譜純,實驗所用其他試劑均為分析純。硅烷化試劑∶雙(三甲基硅烷基)三氟乙酰胺(BSTFA)與三甲基氯硅烷(TMCS)體積比99∶1。
D8 venture型X射線衍射儀:布魯克公司;U-4100型紫外-可見分光光度計:日立公司;TOC-V CPN型總有機碳分析儀:島津公司;7890A/5975C型氣相色譜-質譜(GC-MS)聯用儀:安捷倫公司。
1.2 實驗方法
1.2.1 次氯酸鈉氧化草甘膦
在草甘膦溶液初始質量濃度為16.38 g/L、一定的溶液pH條件下,加入一定量的次氯酸鈉溶液,在轉速200 r/min的磁力攪拌下反應一段時間,每間隔一定時間取樣測定草甘膦的質量濃度,計算草甘膦轉化率。
1.2.2 氧化產物分析
在草甘膦溶液初始質量濃度為50.00 mg/L、溶液pH為9、次氯酸鈉加入量為109 g/L的條件下,反應20 min后用亞硫酸鈉猝滅。取樣冷凍干燥,將所得固體粉末溶于5.00 mL二氯甲烷中,加入0.10 mL硅烷化試劑(BSTFA-TMCS),在60 ℃水浴中硅烷化反應60 min。采用無水硫酸鈉(使用前450 ℃煅燒4 h)脫水,經0.45 μm膜過濾器過濾,氮氣吹掃濃縮為1.00 mL轉移至樣品瓶中,采用GC-MS聯用儀進行分析。
升溫程序:50 ℃保持3 min;以5 ℃/min升溫至150 ℃,保持5 min;以5 ℃/min升溫至250 ℃,保持20 min。進樣口溫度為250 ℃,載氣為高純氦氣,氣流量為1 mL/min。
1.2.3 磷酸銨鎂沉淀法回收磷
次氯酸鈉氧化草甘膦后,磷主要以PO43-的形式存在。磷酸銨鎂沉淀法回收磷的反應見式(1)。

磷酸銨鎂的形成受溶液pH,Mg2+,,的質量濃度及反應時間等因素影響[14]。本實驗采用正交實驗的方法優化磷酸銨鎂生成條件。選取溶液pH、鎂磷摩爾比、氮磷摩爾比、反應時間4個主要影響因素,每個因素設置4個水平,考察指標為磷酸鹽去除率。
采用XRD分析沉淀組成及磷酸銨鎂含量,其中固體樣品生成條件為溶液pH 9.0,鎂磷摩爾比1.2,氮磷摩爾比1.4,氧化時間15 min。
1.3 分析方法
采用X射線衍射儀測定沉淀組成及磷酸銨鎂含量;采用紫外-可見分光光度計測定PO43-質量濃度;采用總有機碳分析儀測定TOC;采用GC-MS聯用儀分析有機物含量。
2 結果與討論
2.1 次氯酸鈉氧化草甘膦的工藝條件優化
2.1.1 次氯酸鈉加入量
在溶液pH為4.9、氧化時間為60 min的條件下,次氯酸鈉加入量對草甘膦轉化率和TOC的影響見圖1。由圖1可見:隨著次氯酸鈉加入量的增加,草甘膦轉化率先逐漸升高;當次氯酸鈉加入量為109 g/L時,草甘膦轉化率達98.2%;繼續增加次氯酸鈉加入量,草甘膦轉化率基本沒有提高。由圖1還可見:在草甘膦轉化率快速升高階段,溶液中的TOC較高;當次氯酸鈉加入量為109 g/L時,TOC為3817.6 mg/L;當次氯酸鈉加入量為259 g/L時,TOC降至2431.2 mg/L;繼續增加次氯酸鈉加入量,TOC變化不大。本實驗次氯酸鈉加入量選擇109 g/L較適宜。

圖1 次氯酸鈉加入量對草甘膦轉化率和TOC的影響
草甘膦轉化率與TOC變化規律的不一致與次氯酸鈉氧化機理有關。次氯酸鈉氧化過程中,首先水解產生ClO-及HClO,由于ClO-價層電子對的空間構型高度不對稱并且中心氯原子具有較大的電子勢,導致其具有較強的獲得電子的能力。另一方面,次氯酸鈉可分解產生具有強氧化性的新生態氧原子[O][15]。次氯酸鈉首先攻擊草甘膦分子中的C—P鍵,含磷基團被氧化為磷酸根;之后,次氯酸鈉才進攻其他有機基團,部分有機碳被轉化為CO2。
2.1.2 溶液pH
在次氯酸鈉加入量為109 g/L、氧化時間為60 min的條件下,溶液pH對草甘膦轉化率的影響見圖2。由圖2可見:當溶液pH為7.0時,草甘膦轉化率為99.5%;增大溶液pH至9.0時,草甘膦轉化率為99.6%;再繼續增大溶液pH,草甘膦轉化率基本保持不變。本實驗選擇溶液pH為9.0較適宜。

圖2 溶液pH對草甘膦轉化率的影響
ClO-在酸性體系中的標準電極電勢1.482 V)高于在堿性體系中但實驗表明,堿性體系中草甘膦轉化率高于酸性體系中的轉化率,這主要是因為酸性條件下次氯酸鈉存在以下反應:

ClO-與次氯酸鈉較Cl2氧化能力更強。溶液pH為2.0時,Cl2為主要存在形態,隨溶液pH增大,次氯酸鈉與ClO-所占比例逐漸增大[16]。式(2)反應的摩爾吉布斯函數表明該反應在標準狀態下可自發進行,且草甘膦氧化為放熱反應,可進一步促進該反應進行。另一方面,溶液中H+可催化分解次氯酸鈉[17],且pH越低反應越劇烈,pH每降低一個單位,有效氯分解速率提高20%左右。堿性體系下,次氯酸鈉自分解速率顯著降低,從而可以提高其有效利用率。
2.1.3 氧化時間
在次氯酸鈉加入量為109 g/L、溶液pH為9.0的條件下,氧化時間對草甘膦轉化率的影響見圖3。由圖3可見:隨著氧化時間的延長,草甘膦轉化率顯著提高;氧化時間為2 min時,草甘膦轉化率為76.8%;氧化時間為10 min時,草甘膦轉化率為96.9%;氧化時間延長至15 min時,草甘膦轉化率進一步提高至97.6%;氧化時間超過20 min后,進一步延長氧化時間,草甘膦轉化率無明顯變化。故本實驗氧化時間控制在20 min即可。
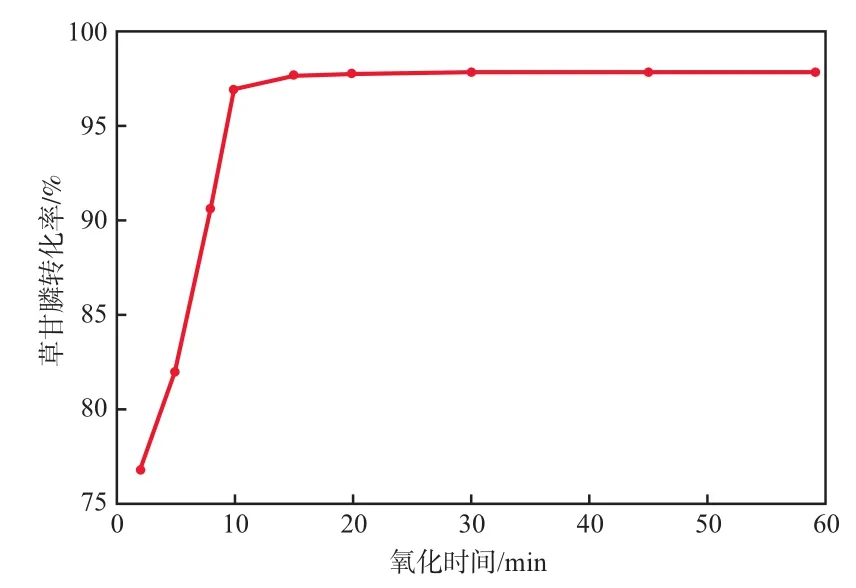
圖3 氧化時間對草甘膦轉化率的影響
2.2 次氯酸鈉氧化草甘膦產物分析
次氯酸鈉氧化草甘膦產物的總離子流圖見圖4。由圖4可見,次氯酸鈉氧化草甘膦的過程中生成了羥基乙酸、磷酸、乙酸等產物。進一步提出了次氯酸鈉氧化草甘膦的主要降解途徑為:C—N鍵斷裂,生成羥基乙酸和乙酸;C—P鍵斷裂生成磷酸。總離子流圖中磷酸濃度最高,乙酸次之,羥基乙酸濃度最低。上述結果表明,C—P鍵斷裂生成磷酸是主要反應,這對于后續磷的回收是有利的。
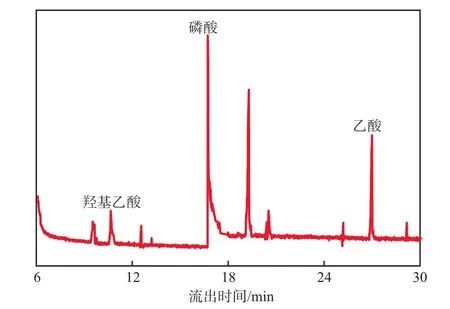
圖4 次氯酸鈉氧化草甘膦產物的總離子流圖
2.3 磷酸銨鎂沉淀法回收磷的正交實驗結果
磷酸銨鎂沉淀法回收磷的正交實驗因素水平見表1。正交實驗結果見表2。其方差分析結果見表3。

表1 磷酸銨鎂沉淀法回收磷的正交實驗因素水平
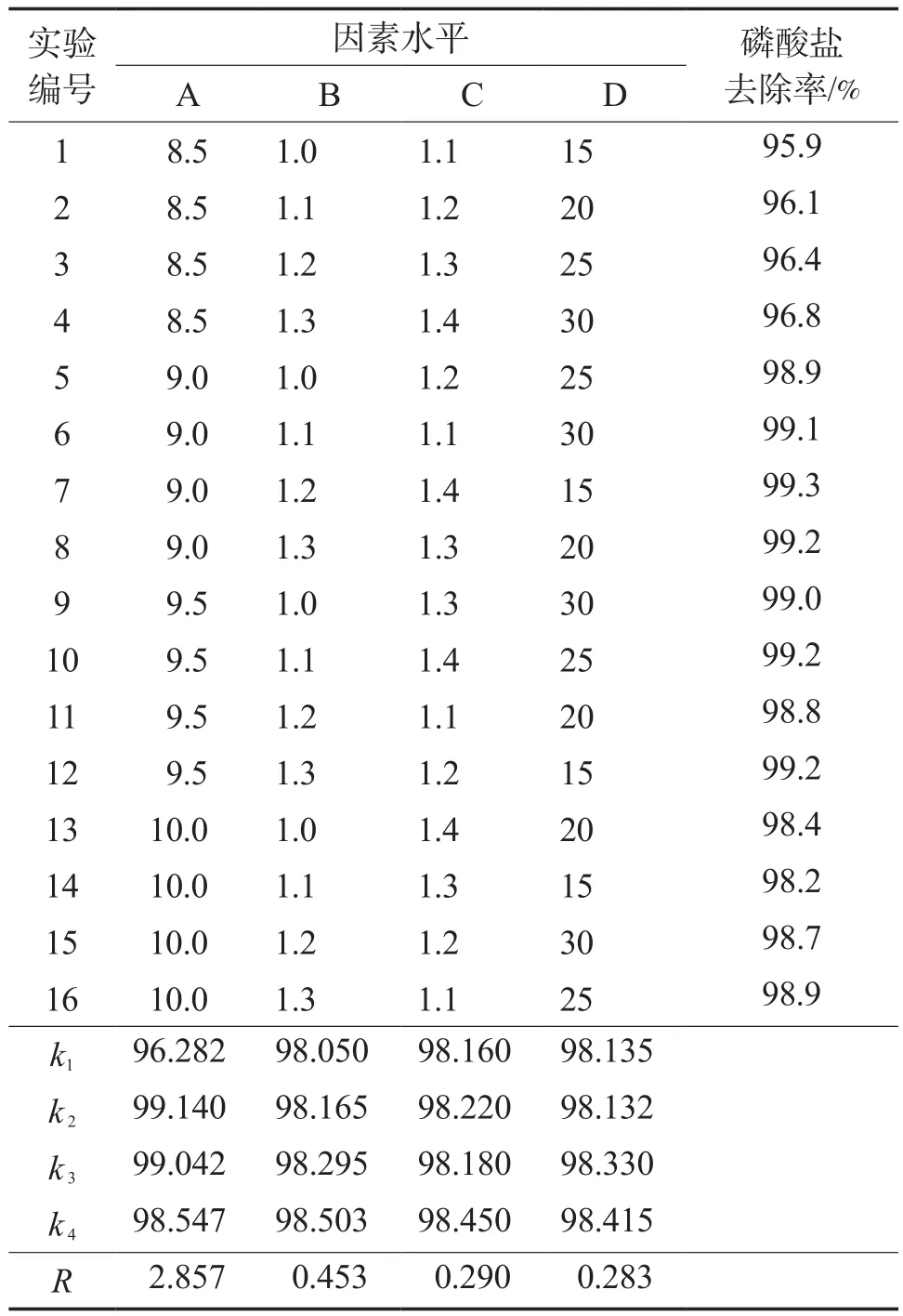
表2 正交實驗結果

表3 正交實驗方差分析表
正交實驗結果表明,對磷回收影響大小的順序為:溶液pH >鎂磷摩爾比>氮磷摩爾比>反應時間。溶液pH對磷回收有顯著影響,其F值與F臨界值分別達到21.8237和7.226,這與前人的研究結果一致[18]。在本研究的實驗條件下,鎂磷摩爾比、氮磷摩爾比對應的F值小于1,表明其對磷回收無顯著影響。正交實驗得到的最佳工藝條件:溶液pH為9.0,鎂磷摩爾比為1.2,氮磷摩爾比為1.4,反應時間為15 min,在此條件下磷酸鹽回收率達99.3%。
采用XRD分析最佳工藝條件下所得固體沉淀的組成及磷酸銨鎂含量,XRD譜圖見圖5。由圖5可見,所得固體沉淀的X射線衍射特征峰與磷酸銨鎂標準譜圖(JCPDS:71-2089)的特征峰吻合,擬合度為87,證實所得固體主要成分為磷酸銨鎂。

圖5 最佳工藝條件下所得固體沉淀的XRD譜圖
3 結論
a)采用次氯酸鈉氧化草甘膦得到磷酸鹽,再采用磷酸銨鎂沉淀法回收磷。次氯酸鈉氧化草甘膦的最佳工藝條件為:次氯酸鈉加入量109 g/L,溶液pH為9.0,氧化時間20 min。
b)次氯酸鈉氧化草甘膦的主要降解途徑為:C—N鍵斷裂生成羥基乙酸和乙酸;C—P鍵斷裂生成磷酸。其中,C—P鍵斷裂生成磷酸是主要反應。
c)正交實驗得到的磷酸銨鎂沉淀法回收磷的最佳工藝條件:溶液pH為9.0,鎂磷摩爾比為1.2,氮磷摩爾比為1.4,反應時間為15 min,在此條件下磷酸鹽回收率達99.3%。
d)所得固體沉淀的X射線衍射特征峰與磷酸銨鎂標準譜圖(JCPDS:71-2089)的特征峰吻合,證實所得固體主要成分為磷酸銨鎂。
[1] 秦龍,姜勝寶,鄭新,等. 草甘膦制劑廢水的資源化利用工藝[J]. 農藥,2015,54(6):411 - 413.
[2] 姜延雄,張悅,陳亞平,等. 草甘膦生產廢水治理技術探討[J]. 環境科技,2015,28(4):76 - 80.
[3] 付雄,劉敏,陳瀅. 污泥灰中磷的濕化學法回收技術研究進展[J]. 化工環保,2017,37(3):276 - 281.
[4] Lu Zijin,Hegemann W. Anaerobic toxicity and biodegradation of formaldehyde in batch cultures[J].Water Res,1998,32(1):209 - 215.
[5] 王曉星,黃應平,鄒雪,等. 草甘膦廢水有機污染物的微生物降解[J]. 環境科學與技術,2010(8):28 -32.
[6] 謝明,徐炎華. 載鐵活性炭對水中草甘膦吸附性能研究[J]. 中國環境科學,2011,31(2): 239 - 244.
[7] McConnell J S,Hossner L R. pH-dependent adsorption isotherms of glyphosate[J]. J Agricul Food Chem,1985,33(6):1075 - 1078.
[8] Huang Yingping,Li Jing,Ma Wandong,et al. Efficient H2O2oxidation of organic pollutants catalyzed by supported iron sulfophenylporphyrin under visible light irradiation[J]. J Phys Chem B,2004,108(22):7263 - 7270.
[9] 申元麗,馬金鋒,趙旭,等. 臭氧氧化降解除草劑草甘膦的實驗研究[J]. 環境科學學報,2011,31(8):1647 - 1652.
[10] Veisi H. Sodium hypochlorite(NaOCl)[J]. Synlett,2007(16):2607 - 2608.
[11] Huang Haiming,Zhang Peng,Zhang Zhao,et al. Simultaneous removal of ammonia nitrogen and recovery of phosphate from swine wastewater by struvite electrochemical precipitation and recycling technology[J].J Cleaner Product,2016,127:302 - 310.
[12] Ishii S K L,Boyer T H. Life cycle comparison of centralized wastewater treatment and urine source separation with struvite precipitation:Focus on urine nutrient management[J]. Water Res,2015,79:88 - 103.
[13] Kataki S,West H,Clarke M,et al. Phosphorus recovery as struvite from farm,municipal and industrial waste:Feedstock suitability,methods and pre-treatments[J]. Waste Manage,2016,49:437 - 454.
[14] 郝曉地,衣蘭凱,王崇臣. 磷回收技術的研發現狀及發展趨勢[J]. 環境科學學報,2010,30(5):897 - 907.
[15] 王萬林. 次氯酸鈉溶液穩定性研究進展[J]. 無機鹽工業,2007,39(9):12 - 14.
[16] Nadupalli S,Koorbanally N,Jonnalagadda S B.Kinetics and mechanism of the oxidation of amaranth with hypochlorite[J]. J Phys Chem A,2011,115(27):7948 - 7954.
[17] 平靜. 次氯酸鈉溶液穩定性的研究[J]. 中國醫院藥學雜志,1995,15(10):455 - 456.
[18] Le Corre K S,Valsami-Jones E,Hobbs P,et al.Agglomeration of struvite crystals[J]. Water Res,2007,41(2):419 - 425.
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
