多系統萎縮合并運動神經元病2例并文獻復習
2018-03-28 08:58:37袁曉利田玉玲
中國現代醫藥雜志 2018年1期
關鍵詞:癥狀
袁曉利 田玉玲
多系統萎縮(Multiple system atrophy,MSA)是一種散發性、快速進展并累及多個神經系統的進行性神經系統退行性疾病,臨床主要以帕金森癥、小腦性共濟失調、自主神經功能障礙和錐體束征等表現為主。根據MSA診斷標準[1],將其分為兩種臨床亞型,一種是以帕金森樣癥狀為突出表現,且對左旋多巴類藥物反應不良的MSA-P型;另一種是以小腦性共濟失調為突出表現的MSA-C型。運動神經元病(Motor neuron disease,MND)是一組病因未明的選擇性侵犯脊髓前角細胞、腦干后組運動神經元、皮質錐體細胞及錐體束的慢性進行性變性疾病,其中肌萎縮側索硬化(Amyotrophic lateral sclerosis,ALS)是其最常見的類型。近年來,隨著影像學技術及其他診斷技術的提高,國外關于多系統萎縮合并運動神經元病的報道及相關研究越來越多,而國內報道很少。現報道多系統萎縮合并運動神經元病2例并結合文獻復習,對多系統萎縮和運動神經元病的發病機制、病理改變、臨床表現、輔助檢查特點及診斷等方面進行探討。
1 臨床資料
1.1 病例1女性,58歲。因“行走困難伴全身乏力3年,加重伴言語含糊1年”于2017年3月10日入院。3年前以走路不穩為首發癥狀,同時伴全身乏力,癥狀呈進行性加重,2年前出現言語含糊、聲嘶、飲水嗆咳、語速變慢、音調變低,近1年上述癥狀逐漸加重,且出現尿失禁、言語含糊到難以被人聽懂、右側肢體無力較左側重,自行抬起費力。對多巴胺能藥物治療反應差。既往肢體發涼多年,大便秘結6~7年,近期加重;否認嗅覺減退及快速眼動期睡眠行為障礙;否認高血壓、糖尿病、心臟病及腦血管病史。患者無特殊個人史、家族史。
體格查體:內科查體無異常。神志清楚,發音困難,吟詩樣語言,有強哭強笑,高級認知功能尚可,查體合作,面具臉,雙側瞳孔等大等圓,直徑約3.0mm,對光反射靈敏,雙眼球各向活動靈活,無復視,雙眼可見水平粗大眼震,雙側眼瞼閉合有力,雙側額紋對稱,示齒鼓腮無力,構音困難,飲水有嗆咳,無吞咽困難,咽反射減弱,軟腭上抬可,伸舌受限不能過齒,可見舌肌纖顫及萎縮。臥床,翻身困難,右側肱二頭肌及雙手骨間肌可見萎縮,四肢肌張力略高,右側上肢肌力3+級,右側下肢肌力3-級,左側上肢肌力5-級,左側下肢肌力4級,右上肢可見震顫,無舞蹈樣動作、手足徐動、肌束顫動等,左側指鼻試驗及跟膝脛試驗欠穩準,輪替試驗差,右側肢體不能配合,右側面部及肢體淺感覺減退,雙側肢體深感覺對稱存在,吸吮反射(+),掌頜反射(+),雙上肢腱反射(++),膝反射(-),跟腱反射(-),雙巴氏征(+),腦膜刺激征(-),體位性血壓不能配合測量,全身皮膚色澤、汗液分布無異常,四肢遠端皮膚冰涼,大便秘結,有小便失禁,余查體不能配合。
輔助檢查:血常規、肝功能、腎功能、血脂、肌酶、電解質、甲功五項、糖化血紅蛋白、葉酸、維生素B12、同型半胱氨酸、凝血系列、風濕系列、腫瘤系列、傳染系列、尿便常規未見明顯異常。頭顱磁共振顯像所見:雙側橋臂可見片狀稍長T1、長T2信號影,小腦及腦干萎縮,可見“十字征”(見圖1、2),小腦腦溝裂增寬,腦回變窄,幕下腦室、腦池擴大;蝶鞍擴大,鞍內可見腦脊液信號,垂體變扁,貼于鞍底。影像診斷:①小腦、腦干萎縮,雙側橋臂異常信號,考慮橄欖橋腦小腦萎縮,雙側半卵圓中心、側腦室旁點狀缺血灶,空蝶鞍;②雙側海馬冠掃未見異常;③頭顱SWI未見明顯異常。自主神經檢測報告提示:自主神經功能偏低。膀胱殘余尿量測定:殘余尿量為55ml,提示殘余尿量增多。頭頸部CTA提示:①左側胚胎型大腦后動脈;②左側頸總動脈分叉處偏心性鈣斑。頸動脈超聲:雙側頸動脈內膜增厚伴斑塊(單發)。腦動脈超聲:腦動脈超聲未見明顯異常。同芯針肌電圖檢查:左胸鎖乳突肌、左右拇短展肌、左股四頭肌外側頭、左右脛前肌肌電圖靜息時見纖顫電位及正銳波,小力收縮見運動單位電位波幅增高、時限增寬、多相波增多,大力收縮募集相減少,呈單純相。右T10、T11脊旁肌肌電圖靜息時見纖顫電位及正銳波,輕收縮可見運動單位電位,參數未測。提示:所檢測肌肉呈神經源性損害。
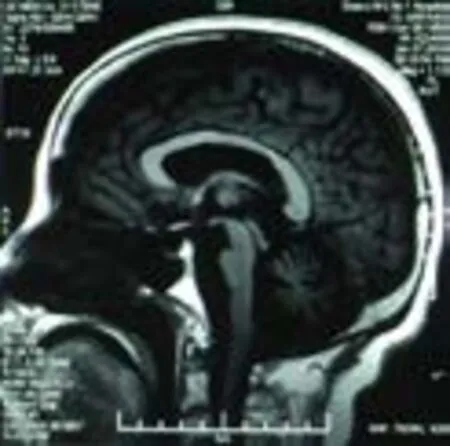
圖1 頭顱MRI T1相示小腦、腦橋萎縮
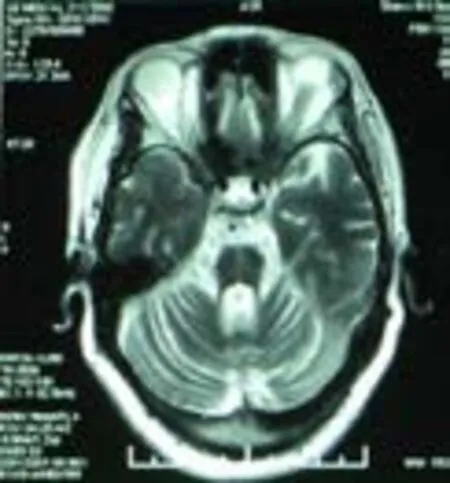
圖2 頭顱MRI T2相示腦橋“十字征”改變
治療經過及效果:入院后給予神經內科系統查體及評估,完善相關化驗及輔助檢查的同時給予鼻飼飲食營養支持,吞咽功能、言語功能、肢體平衡功能的康復鍛煉,考慮有錐體外系受損癥狀,給予口服小劑量多巴絲肼片 1/4片tid(三餐前1h服用),患者錐體外系癥狀改善不明顯,遂增加多巴絲肼片劑量直至劑量達1片tid,患者錐體外系癥狀改善仍不顯著。整個治療過程提示患者對多巴胺能藥物反應差,余治療為靜脈給予丁苯酞注射液抑制自由基及其他改善循環及營養神經等對癥支持治療,患者病情于住院期間未見明顯好轉。
1.2 病例2男性,73歲。因“動作遲緩2年余,加重半年”于2017年9月6日入院。2年前以動作遲緩、肢體僵硬為首發癥狀,同時伴有四肢力弱伴行走拖步、右手靜止及動作時震顫、面部表情減少等癥狀,上述癥狀呈進行性加重,近半年出現言語含糊、發音困難、飲水嗆咳、吞咽困難、流涎、左手輕微震顫、四肢僵硬、翻身及行走困難(需助步器輔助)、雙側大腿根部發沉發困,逐漸生活不能自理并臥床。對多巴胺能藥物治療反應差。既往便秘、尿不盡及夜尿增多10年;高血壓病史15年,口服藥物降壓治療,血壓控制不平穩;2011年因前列腺增生行手術治療,術后小便癥狀未見明顯改善;否認嗅覺減退及快速眼動期睡眠行為障礙;否認糖尿病、心臟病、腦血管病病史。患者無特殊個人史及家族史。
體格檢查:內科查體無異常。神志清楚,言語含糊,發音困難,高級認知功能尚可,查體合作,面具臉,雙側瞳孔等大等圓,直徑約3.0mm,對光反射靈敏,雙眼球各方向活動欠靈活(呈齒輪樣轉動),無眼震及復視,雙側眼瞼閉合有力,雙側額紋及鼻唇溝基本對稱,示齒鼓腮動作遲緩,構音困難,飲水有嗆咳,吞咽困難,咽反射減弱,軟腭上抬力弱,伸舌略受限,基本居中,可見舌肌纖顫,無明顯萎縮。臥床,翻身困難,雙上肢肱二頭肌、肱三頭肌、肱橈肌、骨間肌可見輕度萎縮,雙下肢肌肉萎縮不明顯,雙上肢肌力4級,雙下肢肌力3+級,右側上下肢較左側更差,右手可見靜止性及姿勢性震顫,左手可見靜止性震顫,雙側指鼻試驗及輪替試驗欠穩準,右側為著,跟膝脛試驗不能配合,雙上肢肌張力呈齒輪樣增高,雙側肢體深淺感覺基本對稱,雙上肢羅索里摩征(+),雙上肢腱反射(++),雙下肢腱反射(-),吸吮反射(+),雙側掌頜反射(+),雙巴氏征(+),腦膜刺激征(-),體位性低血壓(+),全身皮膚色澤、汗液分布無異常,四肢遠端皮膚發涼,雙上肢為著,大便秘結,有小便失禁,余查體不能配合。
輔助檢查:血常規、肝功能、腎功能、血脂、肌酶、電解質、甲功五項、糖化血紅蛋白、葉酸、維生素B12、同型半胱氨酸、凝血系列、風濕系列、腫瘤系列、傳染系列、尿便常規未見明顯異常。臥位血壓152/78mmHg,心率75次/min;立位即刻血壓 92/51mmHg,心 率 95次 /min;立 位 3min血 壓106/70mmHg,心率85次/min。頭顱核磁示:①雙側殼核及外側異常信號(見圖3),考慮MSA-P可能,雙側腦室旁、右側半卵圓中心、額葉皮層下多發腔隙性腦梗死,空蝶鞍,大枕大池;②雙側海馬冠掃未見明顯異常;③頭頸部MRA掃描未見明顯異常。24h動態血壓:24h平均血壓141/71mmHg,夜間血壓下降<10%(非勺形曲線),晨起血壓142/80mmHg,監測數值波動規律顯示患者夜間臥位收縮壓最高可達191mmHg,日間立位收縮壓最低可達100mmHg,提示患者自主神經功能調節障礙。膀胱殘余尿量測定:殘余尿量200~250ml,提示殘余尿量增多。同芯針肌電圖檢查:左右拇短展肌、右肱二頭肌、左右脛前肌肌電圖靜息時見纖顫電位及正銳波。左胸鎖乳突肌、左右拇短展肌、左右背側骨間肌Ⅰ、左右脛前肌、右股四頭肌外側頭肌電圖輕收縮見運動單位電位波幅增高、時限增寬、多相波增多。左右拇短展肌、左右背側骨間肌Ⅰ、右肱二頭肌肌電圖大力收縮募集相減少,呈單純相。右T10、T11脊旁肌肌電圖靜息時見纖顫電位及正銳波,輕收縮可見運動單位電位,參數未測。提示廣泛神經源性損害。
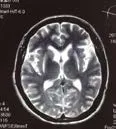
圖3 頭顱MRI示殼核外側緣線樣T2高信號
治療經過及效果:入院后給予神經內科系統查體及評估,完善相關化驗及輔助檢查的同時給予口服小劑量多巴絲肼片 1/2片 qid(三餐前1h及睡前服用),患者帕金森樣癥狀改善不明顯,且體位性低血壓較前波動幅度更大(患者在住院期間出現過兩次臥位變立位暈厥現象),遂給予鹽酸米多君片2.5mg tid治療患者體位性低血壓,監測臥立位血壓結果提示改善不明顯,遂減少多巴絲肼劑量為1/4片tid(三餐前1h服用)后體位性低血壓稍有改善,但帕金森樣癥狀仍改善不明顯,遂后加服鹽酸普拉克索片 1/2片 tid(三餐后1h服用),患者再次出現臥立位血壓波幅增大,且帕金森樣癥狀改善仍不顯著,考慮患者為帕金森疊加綜合征,且對多巴胺能藥物的相關副作用敏感,故未再嘗試其他多巴胺能藥物。整個治療過程提示患者對多巴胺能藥物反應差,余治療為靜脈給予丁苯酞注射液抑制自由基及其他改善循環及營養神經等對癥支持治療,患者病情于住院期間未見明顯好轉。
2 病例分析
病例1患者為中年女性,發病年齡為55歲,病程3年余,起病隱匿,進行性加重。以走路不穩伴全身乏力為首發癥狀,后逐漸出現言語含糊、聲嘶、飲水嗆咳、尿失禁、翻身困難等,呈進行性加重。查體可見面部表情減少、右上肢震顫、翻身困難、四肢肌張力高、輪替試驗差等錐體外系受累體征;雙巴氏征(+)、右側面部及肢體淺感覺減退等錐體束受累體征;四肢遠端皮膚冰涼、便秘、小便失禁等自主神經系統功能障礙體征;雙眼水平粗大眼震、雙側指鼻試驗、跟膝脛試驗(+)等共濟失調為主的小腦癥狀;另有示齒鼓腮無力、構音困難、強哭強笑、飲水嗆咳、舌肌纖顫及萎縮等其他臨床特征;頭顱磁共振提示患者小腦、腦干萎縮,雙側橋臂異常信號,考慮橄欖橋腦小腦萎縮,且可見典型“十字征”;因患者無法站立,故無法監測其臥立位血壓差值,但自主神經功能檢查及膀胱殘余尿量測定提示患者自主神經功能受損,綜上,除腦血管病及遺傳性脊髓小腦共濟失調,根據2008年美國神經病學學會多系統萎縮診斷標準[1],該患者符合MSA的臨床診斷,故診斷為MSA-C型(很可能)。查體可見肌張力增高、病理征陽性、假性延髓麻痹等上運動神經元受累體征;示齒鼓腮無力、伸舌受限不能過齒、舌肌纖顫及萎縮、右側肱二頭肌及雙手骨間肌萎縮、四肢肌力差、右側為著等下運動神經元受累體征,且肌電圖提示延髓、頸髓、胸髓及腰髓神經節所支配的肌肉有進行性失神經和慢性失神經表現。根據2012年中國肌萎縮側索硬化診斷標準[2],可診斷為臨床確診ALS。
病例2患者為老年男性,發病年齡為71歲,病程2年余,同樣為隱匿起病,進展性病程。以動作遲緩、肢體僵硬、四肢力弱、右手靜止及動作時震顫為首發癥狀,逐漸出現面部表情減少、言語含糊、發音困難、飲水嗆咳、吞咽困難、流涎、左手輕微震顫、四肢僵硬、翻身及行走困難、雙側大腿根部發沉發困等癥狀,致患者生活不能自理并臥床。查體可見動作遲緩、靜止性震顫、肌張力增高等錐體外系受累體征;病理征陽性等錐體束受累體征;體位性低血壓、四肢遠端皮膚冰涼、尿便障礙等自主神經系統功能障礙體征;另有發音困難、雙眼球各向活動欠靈活、伸舌略受限、舌肌纖顫、示齒鼓腮動作遲緩、構音困難、飲水有嗆咳、吞咽困難等其他臨床特征;頭顱磁共振提示患者雙側殼核及外側異常信號,考慮為MSA-P型的典型影像學改變殼核裂隙征可能;床旁臥立位血壓測量、24h動態血壓及膀胱大量殘余尿提示患者自主神經功能調節障礙,綜上,除外帕金森病及進行性核上性麻痹,根據相關診斷標準[1],該患者符合MSA的臨床診斷,故診斷為MSA-P型(很可能)。查體同樣可見肌張力增高、病理征陽性、假性延髓麻痹等上運動神經元受累體征;伸舌略受限,舌肌纖顫,雙上肢肱二頭肌,肱三頭肌,骨間肌可見萎縮,四肢肌力差且右側為著等下運動神經元受累體征,肌電圖提示患者延髓、頸髓、胸髓、腰髓神經節所支配的肌肉有進行性失神經和慢性失神經表現。根據2012年中國肌萎縮側索硬化診斷標準[2],可診斷為臨床確診ALS。
3 討論
上述兩例患者的起病形式不同,病例1以小腦性共濟失調癥狀起病,病例2以錐體外系癥狀起病,隨著疾病進展兩例患者均出現運動神經元病同時合并多系統萎縮表現,這在臨床中非常罕見。兩例患者均診斷為多系統萎縮合并運動神經元病。Eisen等[3]曾報道僅有5%的肌萎縮側索硬化患者在疾病過程中會出現帕金森綜合征,而帕金森綜合征又包括原發性帕金森病、多系統萎縮、進行性核上性麻痹等多種疾病,故ALS合并多系統萎縮的比例更小。2000年世界神經病學聯盟修訂的肌萎縮側索硬化診斷標準將ALS進行臨床分型,其中包括散發型ALS、家族遺傳型ALS、ALS疊加綜合征及變異型ALS等幾種亞型,其中ALS疊加綜合征是指ALS與其他神經系統疾病癥狀合并存在,如錐體外癥狀及癡呆等[4],但其臨床特征并未被具體描述。根據上述診斷標準,兩例患者可歸類為ALS疊加綜合征,而此綜合征所包含疾病的病因、發病機制及臨床表現各不相同。多系統萎縮及運動神經元病均屬于神經系統變性疾病,研究證實MSA-P型及MSA-C型的特征性病理改變均為少突膠質細胞內包涵體,其主要成分是一種高度磷酸化的突觸核蛋白[5];而運動神經元病的病因及發病機制目前有多種假說,其確切致病機制迄今未明。
ALS是MND最多見的類型,稱為經典型,大多為散發,也可有家族遺傳,其確切發病機制亦不清楚。上述兩例患者均被診斷為多系統萎縮合并運動神經元病,提示這兩種疾病可能有著共同的發病機制。另外,此兩種疾病共病可能是某一種尚未被認識的疾病,也可能是MSA和MND各自的變異類型。總結國外文獻報道的病例,MSA合并MND有著相似的臨床特征:①單獨的運動神經元病起病年齡比多系統萎縮合并運動神經元病早10歲左右[3~6];②兩種疾病可相繼出現或同時出現[7~9];③多系統萎縮的起病形式多種多樣,且通常對左旋多巴替代治療反應差;④運動神經元病癥狀較突出,且病程與單獨運動神經元病類似。上述兩例患者臨床特點與文獻報道一致,病例1發病年齡比報道的平均發病年齡早14歲,病例2發病年齡比報道的平均發病年齡晚2歲[3],兩例患者均對多巴胺能藥物反應不良,且在住院期間未見明顯肌肉跳動,肉眼可見的肌萎縮也相對不突出,在臨床上容易漏診,需靠同芯針肌電圖提供客觀證據。目前對MSA及MND的治療尚無特異性手段,可根據患者所累及的多個神經系統進行對癥治療,左旋多巴對MSA的錐體外系癥狀可稍有改善但無顯著療效;對自主神經功能受損表現的體位性低血壓,可給予非藥物治療(彈力襪、高鹽飲食等)及藥物治療(鹽酸米多君),但需注意臥位血壓過高;對吞咽困難、言語困難、平衡障礙等可給予鼻飼飲食及康復鍛煉。丁苯酞通過抑制自由基并提高抗氧化酶活性對帕金森疊加綜合征治療有效。MND的治療包括病因治療、對癥治療和各種非藥物治療,目前臨床上應用利魯唑較廣泛,利魯唑具有抑制谷氨酸釋放的作用,服用18個月能延緩病程、延長延髓麻痹患者的生存期,但因其價格昂貴,且疾病進展較快,患者的依從性較差。
MSA和MND同為中樞神經系統變性疾病,MSA合并MND可能并非偶然現象,兩種疾病可能在某一致病因素作用下通過相同的機制使多個中樞神經系統受累,因此在臨床工作中應通過詳細詢問病史及查體,同時結合相關輔助檢查明確MSA患者是否合并MND或MND患者是否合并MSA,以更好地識別并診斷,以免漏診。
1 Gilman S,Wenning GK,Low PA,et al.Second consensus statement on the diagnosis of multiple system atrophy[J].Neurology,2008,71(9):670-676
2 中華醫學會神經病學分會肌電圖與臨床神經電生理學組.中國肌萎縮側索硬化診斷和治療指南[J].中華神經科雜志,2012,45(7):531-533
3 Eisen A,Calne D.Amyotrophic lateral sclerosis,Parkinson's disease and Alzheimer's disease:phylogenetic disorders of the human neocortex sharing many characteristics[J].Can J Neurol Sci,1992,19(S1):117-123
4 Brooks BR,MiHer RG,Swash M,et al.E1 Escorial revisited:revised criteria for the diagnosis of amyotrophic lateral selerosis[J].Amyotroph Lateral Scler Other Motor Neuron Disord,2000,1(5):293-299
5 Shoji M,Harigaya Y,Sasaki A,et al.Accumulation of NACP/alphasynuclein in Lewy body disease and multiple system atrophy[J].J Neurol Neurosurg Psychiatry,2000,68(5):605-608
6 Qareshi AI,Wilmot G,Dihenia B,et al.Motor neuron disease with parkinsonism[J].Arch Neurol,1996,53(10):987-991
7 Zoccolella S,Palagano G,Fraddosio A,et al.ALS-plus:5 cases of concomitant amyotrophic lateral sclerosis and parkinsonism[J].Neurol Sci,2002,23(S2):123-124
8 Klos KJ,Josephs KA,Pafisi JE,et al.Alpha-synuclein immunohistochemistry in two cases of co-occurring idiopathic Parkinson's disease and motor neuron disease[J].Mov Disord,2005,20(11):1515-1520
9 Wiliams TL,Shaw PJ,Lowe J,et al.Parkinsonism in motor neuron disease:case report and literature review[J].Acta Neuropathologic,1995,89(3):275-283
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
